US Activities in Risk Management of Pharmaceutical Products
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: US Activities in Risk Management of Pharmaceutical Products
The mission of the Food and Drug Administration (FDA) is to protect the public health by assuring the safety, efficacy and security of human drugs.
US Activities in
Risk Management of Pharmaceutical Products
INTRODUCTION
The mission of the Food and Drug Administration (FDA) is to protect the public health by assuring the safety, efficacy and security of human drugs. FDA considers risk management to be a continuous process of (1) learning about and interpreting a product’s benefits and risks, (2) designing and implementing interventions to minimize a product’s risks, (3) eval-uating interventions in light of new knowledge that is acquired over time and (4) revising interventions when appropriate.
The
avoidance of serious harm is the most commonly asserted justification for
public health regulation (Gostin, 2000). Pharmaceutical risk management is the
overall and continuing process of minimizing a drug’s risks throughout its life
cycle to optimize its benefit/risk balance. Risk information emerges
continuously throughout this life cycle, during both the investigation and
marketing phases through both labelled and off-label uses.
This
chapter will provide an overview of recent US regulatory activities in risk
management and its evolving role in post-marketing surveillance of
phar-maceutical products.
In
May 1999, the Task Force on Risk Manage-ment issued its report to the
Commissioner of FDA. Traditionally, FDA has filled several
important roles in minimizing the risks associated with medical product use by
establishing and enforcing product quality standards intended to prevent
defective prod-ucts from reaching the market. Furthermore, this report
challenged the traditional model by its care-ful analysis of the challenges
faced in managing risks within the context of the broader healthcare delivery
system (Figure 43.1).
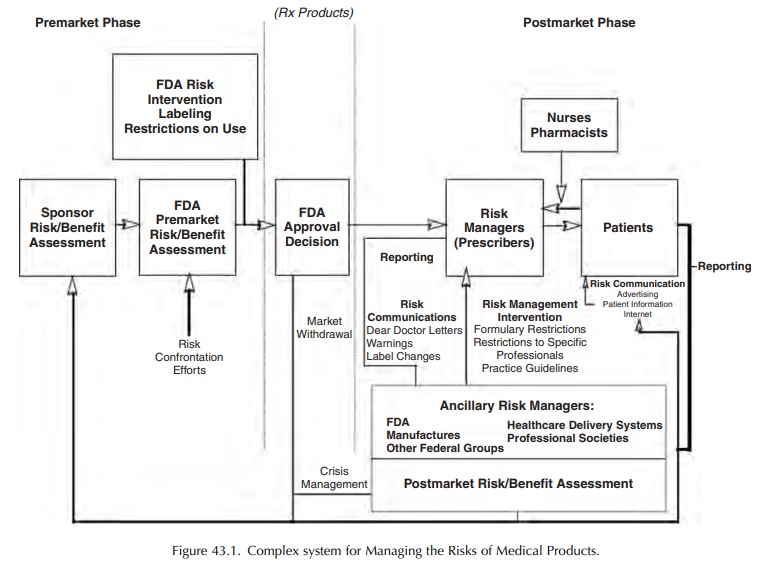
FDA
evaluates the safety profiles of drugs available in the United States using a
variety of tools and disciplines throughout the life cycle of the drugs. Under
US regulations,3 manufacturers of approved drug and biologic
products are required to promptly report all adverse drug experience
information obtained or otherwise received by the manu-facturer from any
source, foreign or domestic, including information derived from commercial
marketing experience, post-marketing epidemiologi-cal/surveillance studies, reports
in the scientific liter-ature and unpublished scientific papers. FDA also
accepts reports directly from healthcare providers and consumers. Currently,
the agency’s adverse event database has over 3.5 million reports with
increasing numbers reported annually (Figure 43.2).
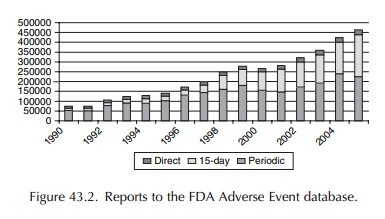
This
system of post-marketing surveillance report-ing [the adverse event reports
system or adverse event reporting system (AERS)] and risk assessment programmes
serves to identify adverse events that did not appear during the drug
development process. The successful implementation of electronic submissions is
a high priority for the center. Further improve-ments in this system include
electronic submission of adverse drug reports that will result in more timely
receipt and evaluation of adverse event reports at considerable cost savings
both to FDA and to those submitting the reports. Data mining provides an
important tool in facilitating signal detection of the more than three million
reports in this database.
Related Topics
