Sterility Testing
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Sterilization Procedures And Sterility Assurance
A sterility test is essentially a test which assesses whether a sterilized pharmaceutical or medical product is free from contaminating microorganisms by incubation of either the whole or a part of that product with a nutrient medium.
STERILITY TESTING
A sterility
test is essentially a test which assesses whether a sterilized pharmaceutical or medical product
is free from contaminating
microorganisms by incubation of
either the whole or a part of that product
with a nutrient medium. It thus
becomes a destructive test and is of
questionable suitability for testing large,
expensive or delicate products or equipment. Furthermore, by its very nature such a test is a statistical process in which
part of a batch is sampled and the chance
of the batch being passed for use then depends
on the sample passing the sterility test. Random sampling
should be applied to products that have been processed and filled aseptically. With products
sterilized in their
final containers, samples should be taken from the potentially coolest
or least sterilant-accessible part of the load.
A further
limitation is that
which is inherent in a procedure intended to demonstrate a negative. A sterility test is intended to demonstrate that
no viable organisms are present, but failure
to detect them could simply
be a consequence
of the use of unsuitable media or inappropriate cultural conditions. To be certain that
no organisms are present
it would be necessary to use a universal culture medium suitable for
the growth of any possible
contaminant and to incubate
the sample under
an infinite variety of conditions. Clearly, no such medium
or combination of media are available and, in practice, only media capable of supporting non-fastidious bacteria,
yeasts and moulds are employed. Furthermore, in pharmacopoeial tests,
no attempt is made to detect viruses,
which on a size basis, are the organisms most likely to pass through
a sterilizing filter. Nevertheless,
the sterility test
does have an important application in monitoring the
microbiological quality
of filter-sterilized, aseptically filled products and does offer a final check
on terminally sterilized articles. In the UK, test procedures laid down by the European
Pharmacopoeia
must be followed; this provides details
of the sample sizes to be adopted
in particular cases. The
principles of these tests
are discussed below.
a)
Methods
Three alternative methods are available when conducting sterility tests:
•
The direct inoculation method involves introducing test samples
directly into nutrient media. The European
Pharmacopoeia recommends two media:
(1) fluid mercaptoacetate medium (also known as fluid thioglycollate medium), which
contains glucose and
sodium mercaptoacetate
(sodium thioglycollate) and is particularly suitable for the cultivation of anaerobic
organisms (incubation
temperature 30–35 °C); and (2) soyabean casein digest medium (also known as tryptone
soya broth), which will support the growth of both aerobic bacteria (incubation temperature 30–35 °C) and fungi
(incubation temperature 20–25
°C). Other media
may be used provided that they can
be shown to be suitable alternatives. Limits are
placed upon the
ratio of the weight
or volume of added sample
relative to the volume of culture medium so as to avoid
reducing the nutrient properties of the
medium or creating unfavourably high osmotic pressures within it.
•
Membrane filtration is the
technique recommended by most pharmacopoeias and, consequently,
the method by which the
great majority of products are examined. It
involves filtration of fluids through
a sterile membrane filter (pore size ≤0.45
μm), any microorganism present being retained on the surface
of the filter. After
washing in situ, the filter is divided
aseptically and portions
are transferred to suitable culture media which are then incubated at the appropriate temperature for the required period of time.
Water-soluble solids
can be dissolved in a suitable diluent and processed in this way and oil-soluble products may be dissolved in a suitable
solvent, e.g. isopropyl myristate.
•
A sensitive
method for detecting low levels of contamination in intravenous infusion fluids involves
the addition of a concentrated culture
medium to the fluid in its
original container, such that the
resultant mixture is equivalent to single strength culture
medium. In this way, sampling of the entire volume
is achieved.
With the techniques
discussed above, the media employed should previously have been
assessed for nutritive (growth-supporting) properties and a lack
of toxicity using specified organisms. It must
be remembered that any survivors of a sterilization process
may be damaged and thus must be given
the best possible
conditions for growth.
As a precaution against
accidental contamination, product testing must
be carried out
under conditions of strict asepsis using,
for example, a laminar airflow
cabinet to provide
a suitable environment.
The European Pharmacopoeia indicates that it is necessary to conduct
control tests that
confirm the adequacy of the facilities by sampling of air and surfaces and carrying out tests using
samples ‘known’ to be sterile
(negative controls). In reality, this means samples
that have been subjected to a very reliable sterilization process, e.g. radiation, or samples that have been subjected to a sterilization procedure more
than once. In order to minimize
the risk of introducing contaminants from the surroundings or from
the operator during
the test itself,
isolators are
often employed which physically separate the operator from the materials under test.
b) Antimicrobial Agents
Where an antimicrobial agent
comprises the product
or forms part of the product, for example as a preservative, its activity must be nullified in some way during sterility testing so that
an inhibitory action
in preventing the growth of any contaminating microorganisms is overcome. This is achieved by the following methods.
i) Specific inactivation
An appropriate inactivating (neutralizing) agent (Table 21.8) is incorporated into
the culture media.
The inactivating agent must be non-toxic to microorganisms, as must
any product resulting from an interaction of the inactivator
and the antimicrobial agent.
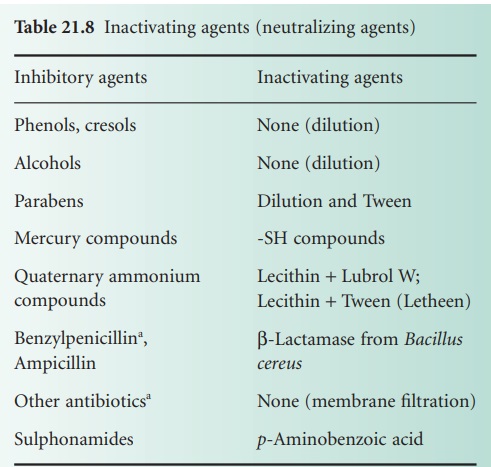
Although Table 21.8 lists only benzylpenicillin and ampicillin as being inactivated by β-lactamase (from
B. cereus), other β-lactams may also be hydrolysed by βlactamases. Other antibiotic-inactivating enzymes
are also known and have been considered as possible inactivating agents, e.g. chloramphenicol acetyltransferase (inactivates chloramphenicol) and enzymes that modify aminoglycoside antibiotics.
ii) Dilution
The antimicrobial agent is diluted
in the culture medium to a level at which it ceases
to have any activity, for example phenols, cresols and alcohols. This method applies
to substances with a high
dilution coefficient, η.
iii) Membrane filtration
This method
has traditionally been used to overcome the activity of antibiotics for which there
are no inactivating agents, although it could be extended to cover other products if necessary, e.g. those containing preservatives for which no specific or effective inactivators are available. Basically, a solution of the product
is filtered through a hydrophobic-edged membrane filter
that will retain
any contaminating
microorganisms. The membrane is washed in situ to
remove any traces
of antibiotic adhering to the membrane and is then
transferred to appropriate culture media.
c)
Positive Controls
It is essential to show that microorganisms will actually
grow under
the conditions of the test.
For this reason positive controls have to be carried
out; in these,
the ability of small numbers
of suitable microorganisms to grow in media
in the presence of the sample
is assessed. The microorganism used for positive control tests with
a product containing or comprising an antimicrobial agent must, if at all possible, be sensitive to that agent,
so that growth of the organism
indicates a satisfactory inactivation, dilution or removal of the agent.
The European Pharmacopoeia suggests the use of designated strains of Staphylococcus aureus, Bacillus subtilis and Pseudomonas aeruginosa as appropriate aerobic
organisms, Clostridium sporogenes as an anaerobe and Candida albicans or Aspergillus niger as fungi.
In practice, a positive control
(medium with added test sample) and a negative control
(medium without it) are inoculated simultaneously, and the rate and
extent of growth arising in each should be similar. However, the negative control
without the test sample,
is, in effect, exactly the same as the growth
promotion control that is also described in the test procedure, so, for
the organisms concerned, it is not necessary to do
both.
All the
controls may be conducted either
before, or in parallel with, the test itself,
providing that the same
batches of media are used for both.
If the controls are carried out in parallel with the tests
and one of the controls gives an unexpected result,
the test for sterility may be declared invalid,
and, when the problem is resolved,
the test may be repeated.
d) Specific Cases
Specific details
of the sterility testing of parenteral products, ophthalmic and other non-injectable
preparations, and surgical
sutures will be found in the European
Pharmacopoeia. These procedures cannot conveniently be applied to items like surgical dressings
and medical devices because
they are too big. In such cases
the most convenient approach
is to immerse the whole object in culture medium in a sterile flexible
bag, but care must be taken to ensure
that the liquid
penetrates to all parts and surfaces of the material.
e)
Sampling
A sterility test attempts to infer the state (sterile
or nonsterile) of a batch from the results of an examination of part of a batch,
and is thus
a statistical operation. Suppose that p represents the proportion of infected containers in a batch
and q the proportion of non-infected containers,
then, p +
q = 1 or q = 1 − p.
Suppose also
that a sample
of two items is taken
from a large
batch containing 10% contaminated containers. The probability of a single
item taken at random being contaminated is p = 0.1
(10% = 0.1), whereas the probability of such an item being
non-contaminated is given by q = 1 − p =
0.9. The probability of both items
being contaminated is p2 = 0.01, and of both items being non-contaminated, q2 = (1 − p)2 = 0.81. The probability
of obtaining one contaminated item and one non
contaminated item is 1 − (0.01 + 0.81) = 0.18 = 2pq.
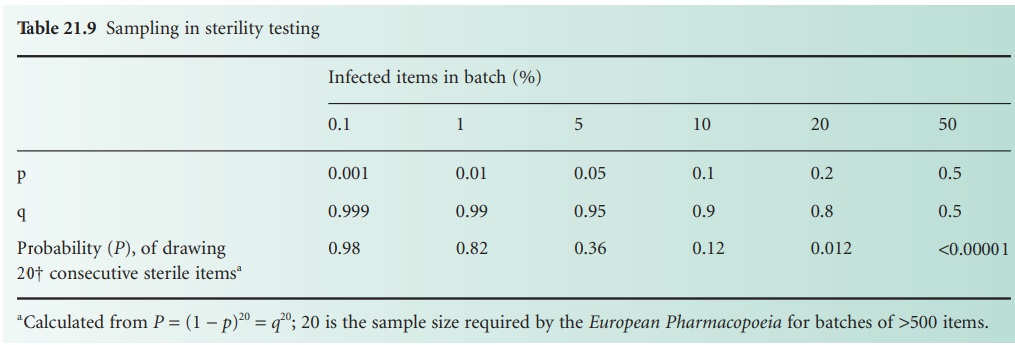
In a sterility test
involving a sample
size of n containers, the probability p of
obtaining n consecutive ‘steriles’ is given by qn = (1 − p)n. Values for
various levels of p (i.e. proportion of infected containers in a batch)
with a constant sample size are given in Table 21.9, which shows that the test cannot
detect low levels
of contamination. Similarly, if different sample sizes are
employed (also based on (1 − p)n) it can be shown that
as the sample
size increases, the probability of the batch being passed
as sterile decreases.
It can be seen from the above that a sterility
test can only show
that a proportion of the
products in a batch is sterile. Thus, the correct
conclusion to be drawn from a
satisfactory test result
is that the batch has passed the sterility test not that the batch is sterile.
f)
Retests
Under certain circumstances a sterility test may be repeated, but the only justification for repeating the test
is unequivocal evidence
that the first
test was invalid;
a retest cannot be viewed as a second
opportunity for the batch to pass
when it has
failed the first
time. Circumstances that may justify
a retest would
include, for example, failure of the air filtration system in the testing facility which might have permitted
airborne contaminants to enter
the product or media during
testing, non-sterility of the media used
for testing, or evidence that
contamination arose during testing
from the operating
personnel or a source other
than the sample
under test.
Related Topics
