Solved Problems on Stereochemical and Conformational Isomerism
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Stereochemical and Conformational Isomerism
Questions and answers, Solved Problems on Stereochemical and Conformational Isomerism - Organic Chemistry
PROBLEMS
6.1. Indicate the chiral
centers in the following molecules and give the relative configuration (R,S) of
each:
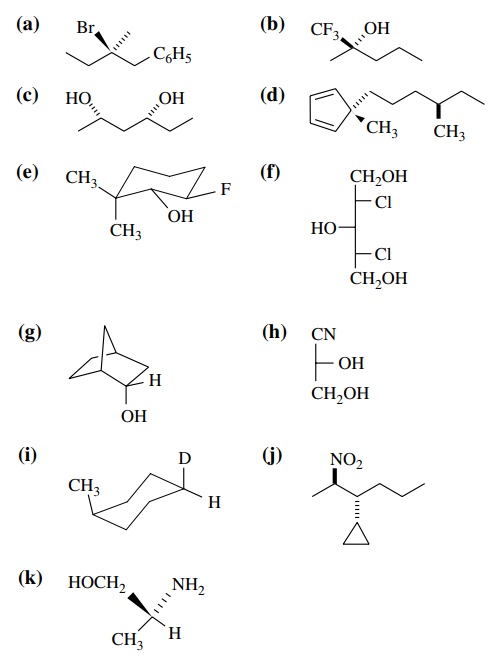
Answer:
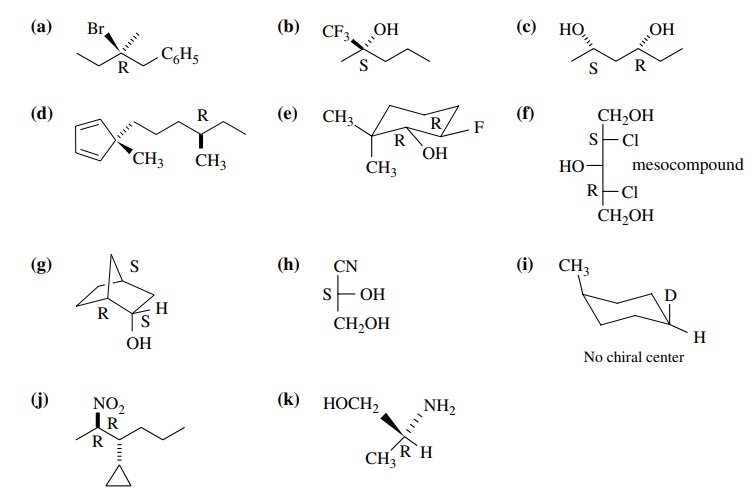
6.2. The following
structures are representations of 3-fluoro-2-phenyl-2-pentanol. Give the
stereochemical relationship of each structure to
(2R,3R)-3-fluoro-2-phenyl-2-pentanol:
Answer:
(a)
2R, 3R
(b)
2S, 3R diastereomers
(c)
2R, 3R identical
(d)
2S, 3S enantiomer
(e)
2R, 3S diastereomer
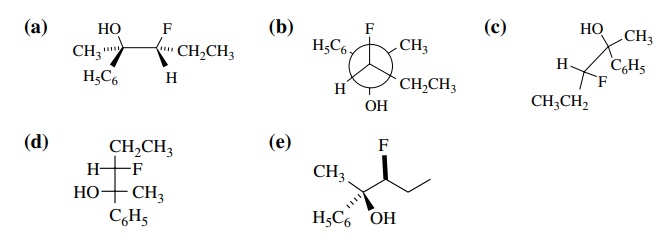
6.3. (a) Draw the
four stereoisomers of 4-methyl-2-hexanol and give the relationship of each to
the others.
(b) Draw all the
stereoisomers of 3-bromo-4-methylhexane, give the R,S designation of each
chiral center, and give the relationship of each to the others.
(c) Using Fischer
projections, draw all of the stereoisomers of 2-fluoro-3-methyl-1,4-pentanediol
and give the relationship of each to the others.
(d) Draw all of the stereoisomers of 1,4-diphenyl-1,4-dibromobutane, give the R,S designation of each chiral center, and give the relation-ship of each to the others.
Answer:
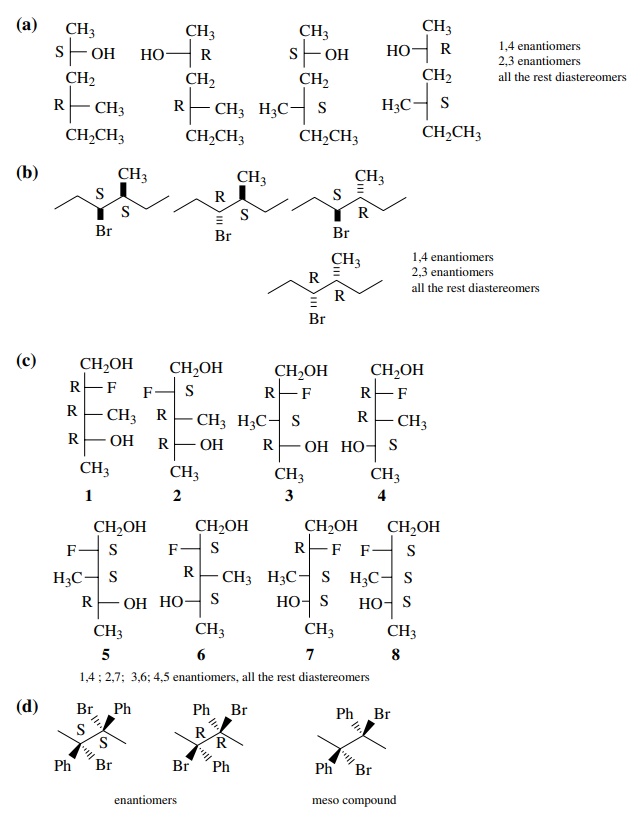
6.4. Give the
stereochemical relationship between the following pairs of com-pounds:
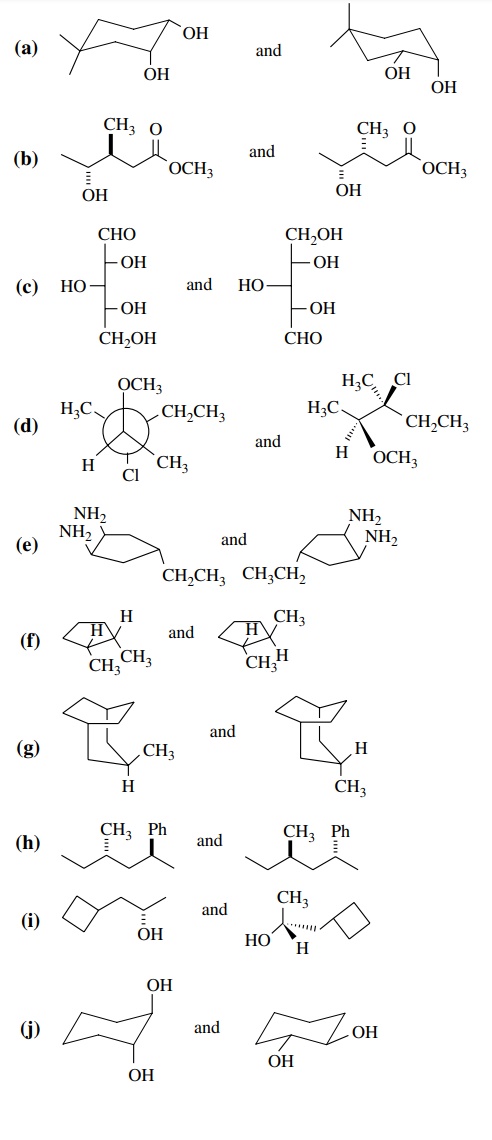
Answer:
(a) Both 1S, 2R, conformational isomers
(b)
Syn – anti, diastereomers
(c)
Enantiomers
(d)
Enantiomers
(e)
Identical
(f)
Diastereomers
(g)
Conformational diastereomers
(h)
Enantiomers
(i)
Enantiomers
(j)
Conformational isomers
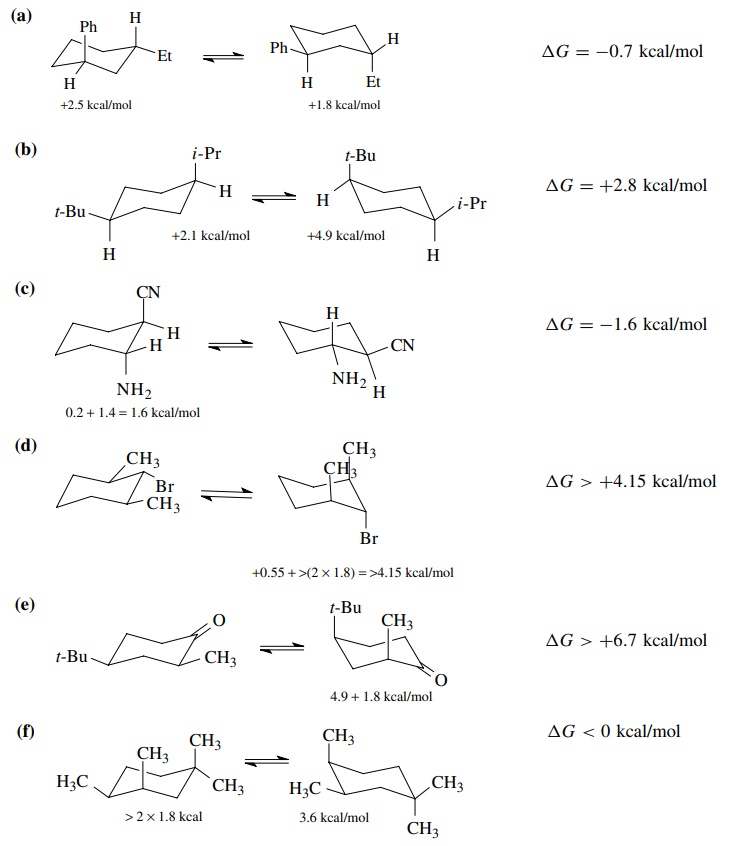
6.5. For the
following compounds, show two chair conformations, indicate which is more
stable, and give an estimate of the energy difference between the two:
(a) trans-1-ethyl-3-phenylcyclohexane
(b) cis-1-(tert-butyl)-4-isopropylcyclohexane
(c) trans-2-amino-1-cyanocyclohexane
(d) (2R,6S)-1-bromo-2,6-dimethylcyclohexane
(e) cis-4-(tert-butyl)-2-methylcyclohexanone
(f) cis-1,1,3,4-tetramethylcyclohexane
Answer:
6.6. Estimate the difference in energy between the chair conformations of trans-2-methoxycyclohexanol. The actual value is about 3.1 kcal/mol. Can you explain this?
Answer:
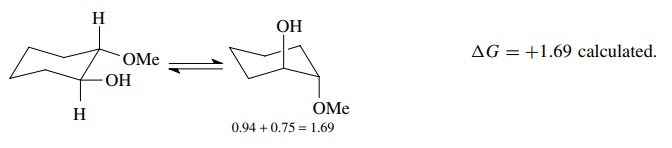
Because of H bonding in the diequatorial form, ΔG includes
the energy required to break the H bond. Thus the actual ΔG is greater than
that calculated.
6.7. Show all of the
staggered conformers of 2,3-dimethylbutane and estimate the energy differences
between them.
Answer:
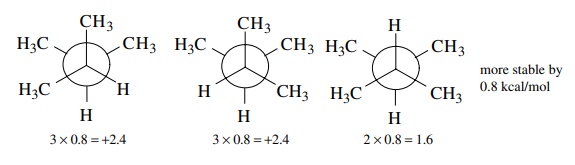
6.8. The Beckman
rearrangement could occur by either a stepwise or a con-certed mechanism.
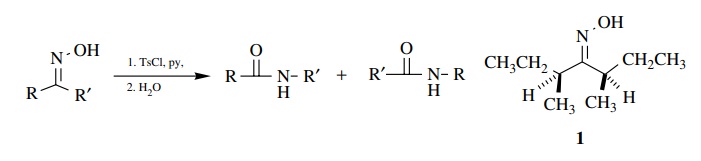
(a) Show both
mechanisms using curved-arrow notation.
(b) Suppose you had
made oxime 1.
1. Would it rotate
plane-polarized light?
2. Label the
configurations of the chiral centers in 1.
3. Show how 1 could be used to help distinguish the mechanisms you have given.
Answer:
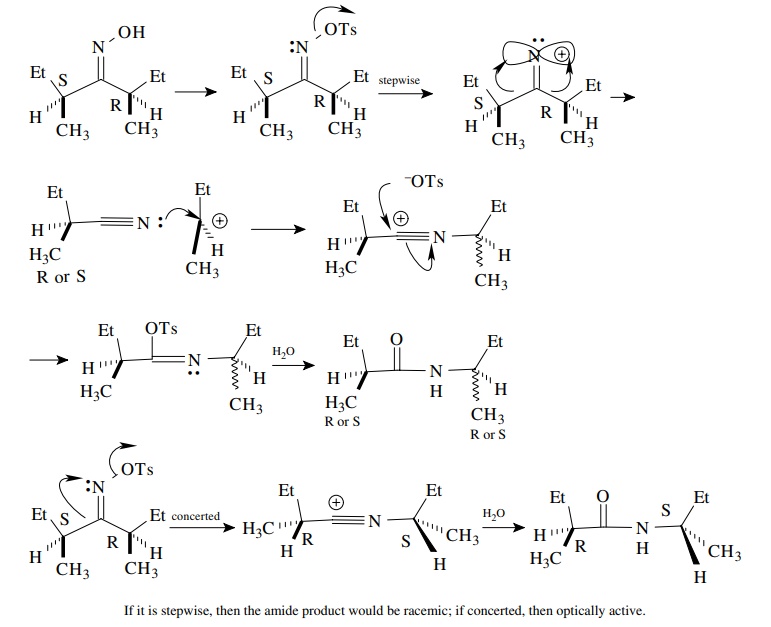
6.9. Explain why (1S,3R)-3-tert-butylcyclohexyl tosylate undergoes E2 elimi-nation with potassium tert-butoxide very slowly while the (1R,3R) reacts much more rapidly.
Answer:
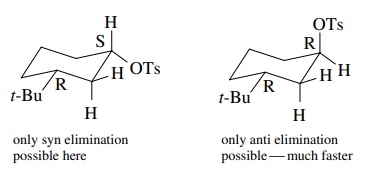
6.10. The reaction of cis-2-pentene with iodine azide (IN3) in dichloromethane gives (2S,3S)-3-azido-2-iodopentane and (2R,3R)-3-azido-2-iodopentane but not any other diastereomers. What is the stereochemistry of the addi-tion and give a curved-arrow mechanism to account for it.
Answer:
6.11. The reaction of trans-2-hexene with aqueous peracetic acid gives (2S,3R)-2,3-hexane diol and (2R,3S)-2,3-hexanediol but not any other diastere-omers. What is the stereochemistry of the addition?
Answer:
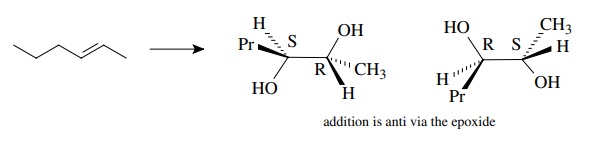
6.12. Heating (2S)-3-methyl-3-phenyl-2-butyl tosylate in ethanol leads to skeletal rearrangement and the formation of (3S)-2-ethoxy-2-methyl-3-phenylbutane. What does this information tell you about the stereoelectronic course of the skeletal rearrangement?
Answer:
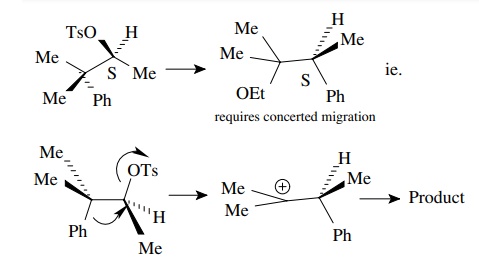
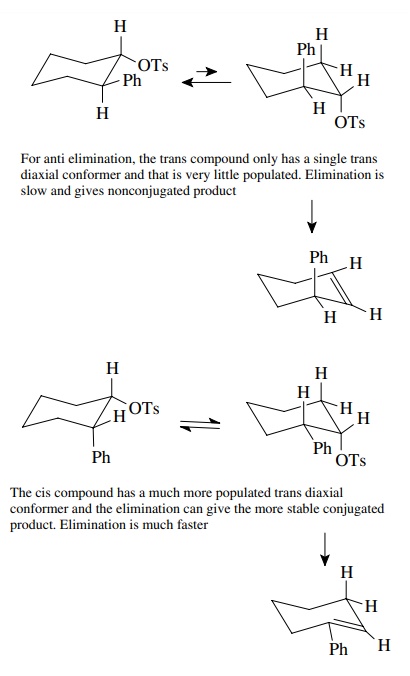
6.13. Treatment of trans-2-phenylcyclohexyl tosylate with potassium tert-butoxide gives mainly 3-phenylcyclohexene in a fairly slow process, whereas under the same conditions cis-2-phenylcyclohexyl tosylate gives 1-phenylcyclohexene in a much shorter reaction time. Explain this difference.
Answer:
6.14. What do the
following strain energies suggest about the origin of strain in three-membered
rings?
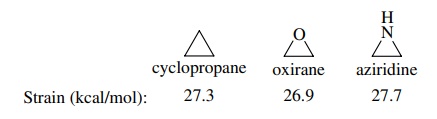
Answer:
The origins of strain in three-membered rings could be both
angle strain and torsional strain. Since lone pairs are effectively smaller
than bonds to hydrogen, replacing the C–H bonds in cyclopropane by one lone
pair in aziridine or by two lone pairs in oxirane should reduce torsional
strain. Since these changes do not change the strain, it is clear that strain
in three-membered rings is due entirely to angle strain.
6.15. Although
cyclobutane is a puckered molecule (by about 25◦ ), its oxygen analog oxetane is virtually flat. Give a
rationale for this behavior.
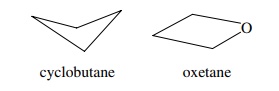
Answer:
Puckering in four-membered rings is due to the molecule
relieving torsional strain. In doing so, some additional angle strain is
introduced but the net result is the most stable structure. Replacing the C–H
bonds by lone pairs which are smaller than C–H bonds lessens the torsional
strain so the molecule flattens to reduce angle strain.
6.16. The strain
energy of spiropentane (62.5 kcal/mol) is more than twice that of cyclopropane
(27.3 kcal/mol). Suggest an explanation.
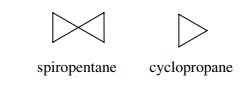
Answer:
Normally the exocyclic bonds (i.e., the C–H bonds) of
cyclopropane are greater than 109◦ and in fact approach 120◦ . This is because
the ring bonds have greater p character to accommodate the smaller angle and
the external bonds have greater s character. The greater s character causes the
angles to be greater than 109◦ . Adding a spiro ring now forces
the spiro carbon to have exocyclic bond angles of less than 109◦ . This adds
increased strain to the system such that the strain is greater than to single
cyclopropyl rings (2 ×
27.3 kcal/mol = 54.6 kcal/mol).
6.17. Based on the
properties of the cyclohexane ring, which of these isomers is predicted to have
a larger dipole moment? Explain your choice.
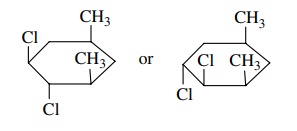
Answer:
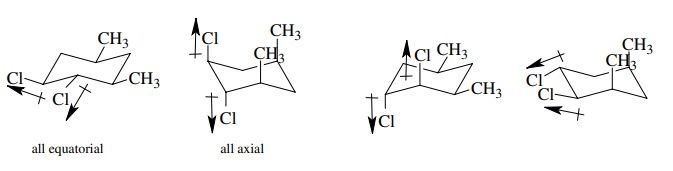
Inspection of the two sets of chair structures reveals that
in one compound the all-equatorial conformer is overwhelmingly favored. In the
other compound both chair structures have comparable energies so both will be
populated significantly. In the all-equatorial isomer, the carbon – chlorine
bond dipole moments reinforce one another leading to a large molecular moment.
In the other compound the chlorines are both equatorial part of the time but
part of the time they are trans diaxial where the carbon – chlorine bond dipole
moments tend to cancel one another. Thus the average dipole moment of these two
conformations will be less than the first compound, which exists virtually
completely in the all-equatorial conformer.
6.18. Draw the conformational isomers of cis-1,2-dimethylcyclohexane and cis-3,4-dimethylcyclohexanone. While the cyclohexane conformers are of equal energy, the cyclohexanone conformers are not. Indicate which con-former is favored and explain why.
Answer:
In the cyclohexane there is one axial methyl in either
conformation; thus the two conformations are of equal energy and will be
equally populated. Conformer 1 has a
1,3 diaxial methyl – proton interaction and a 1,3 interaction between the
methyl group and the carbonyl group. Conformer 2, on the other hand, has two 1,3 diaxial methyl – proton interactions.
Since the carbonyl group is somewhat smaller than a CH2 group,
conformer 1 has slightly smaller 1,3
diaxial interactions and therefore will be favored slightly.
6.19. Addition of
osmium tetroxide to norbornene 2 followed by reductive cleavage with sodium
sulfite gives the exo,exo diol 3. The same reac-tion sequence carried out on
7,7-dimethylnorbornene 4 gives endo,endo diol 5. From these results deduce the
mechanism of the addition and facial selectivity for these two substrates.
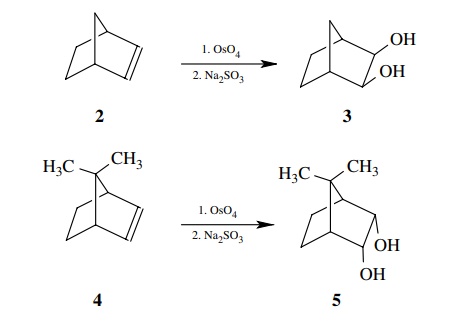
Answer:
The fact that only cis product is produced from either 2 or 4 means that both oxygens in the product come from the osmium
reagent. This could result from either a concerted, 3 + 2 cycloaddition of OsO4
to the double bond or a sequential addition of one then another oxygen to the
same side of the double bond.
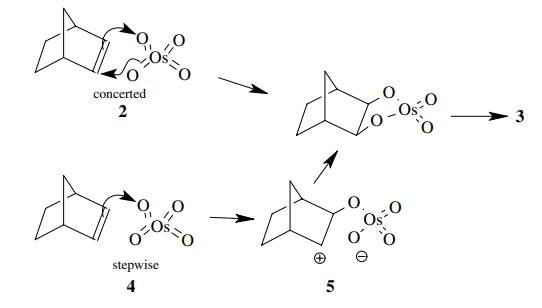
Comparison of 2
and 4 shows that the facial
preference is for exo attack. Only when the exo face is blocked by a methyl
group does the reagent attack the endo face.
6.20. Treatment of E-1-phenyl-2-butene 5 with I2
and silver benzoate (1 : 2) followed by saponification gives an equal mixture
of [2S,3R]-1-phenyl-2,3-butanediol and [2R,3S]-1-phenyl-2,3-butanediol.
Treatment of 5 with I2 and silver acetate in the presence of water
followed by saponification gives an equal mixture of [2R,3R]-1-phenyl-2,3-butanediol
and [2S,3S]-1-phenyl-2,3-butanediol. Determine the stereochemistry for these
two pro-cesses. Can you account for the difference mechanistically?
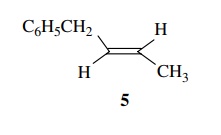
Answer:
Knowing
that ultimately two hydroxyl groups are added to each end of the double bond,
one can construct the possible isomers easily.
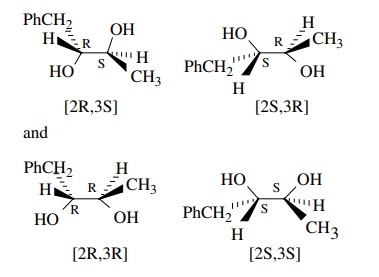
By
inspection one can see that the the 2R,3S and 2S,3R isomers are enantiomers and
the 2R,3R and 2S,3S pair are enantiomers. As they are drawn, one can see the
original E stereochemistry of the double bond. Thus the first pair comes from a
stereospecific trans addition across the double bond while the second pair
comes from a stereospecific cis addition. Clearly the stereochemistry of the
double bond is maintained throughout the addition. Since iodine is the
electrophile, a bridged iodonium ion is likely responsible. The initial
addition gives only a single pair of enantiomers since the only nucleophile is
benzoate. Also noted is that iodine is not in the product so something must
replace the iodine. The silver ion would help to remove the iodine. Since
iodine would cause a trans addition and the product has oxygens added trans,
there must be a replacement of iodine by an oxygen ligand with retention of
configuration at the iodine center. Neighboring group participation is
responsible.
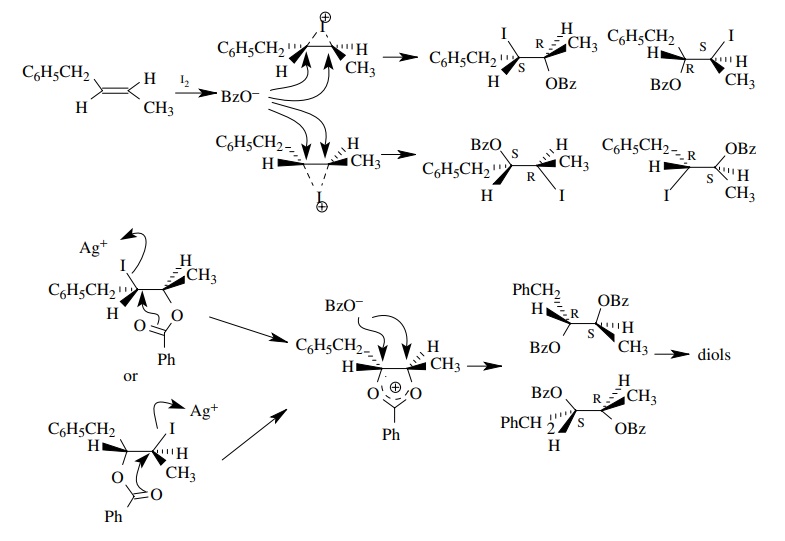
The
other set of enantiomers must result from inversion of configuration. The pres-ence
of water must prevent the benzoyl group from acting as a neighboring group.
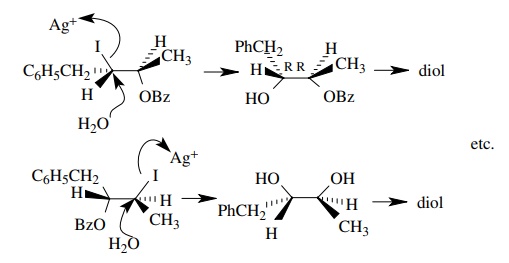
6.21. Treatment of
[4S]-4-t -butyl-1-methylcyclohexene with borane – THF followed by
oxidation with H2O2 – NaOH gives a mixture of [1S, 2S,5S]-5-t
-butyl-2-methylcyclohexan-1-ol and [1R,2R,5S]-5-t -butyl-2-methylcyclohexan-1-ol and no
other diastereomers. There are two steps in this process—addition to give an
organoborane and oxidation which cleaves the carbon – boron bond to an alcohol.
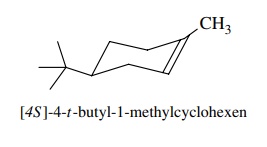
(a) Is the overall
process stereospecific?
(b) What are the
stereoselectivities of each step (mode of addition and stereochemistry of
cleavage) which are consistent with the observed products?
(c) It is known that the oxidation reaction occurs with retention of configuration. What then must be the stereochemistry of the hydrob-oration step?
Answer:
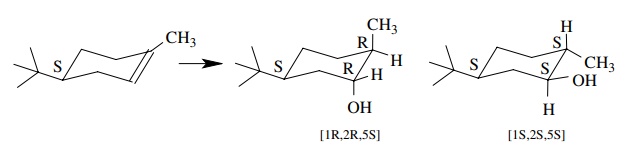
(a) From the products it is clear that a hydrogen and a
hydroxyl have added in a syn fashion to either face of the olefin.
(b)
Since a hydrogen and a boron add in the first step and then the boron ligand is
converted to an oxygen ligand in the second step, the possibilities are (a)
mode of addition syn–anti and (b)
stereochemistry of cleavage retention–inversion.
Picking only the lower face for the initial addition step, the four
stereochemical possibilities are
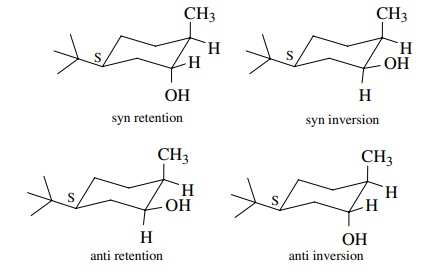
(c)
Since the actual product has the hydroxyl group and methyl groups cis to one
another, the syn retention and anti inversion sequences are the only possible
ones. If it is known that cleavage occurs with retention, then the
hydroboration reaction must occur by syn addition across the double bond.
6.22. Treatment of [2R,3R]-2,3-dibromo-3-methylpentane with Zn gives (Z)-3-methyl-2-pentene as the only product. What is the stereochemistry of the reduction? Based on this result could the reaction of a trans olefin with Br2 and then Zn be used as a way to convert trans olefins to cis olefins?
Answer:
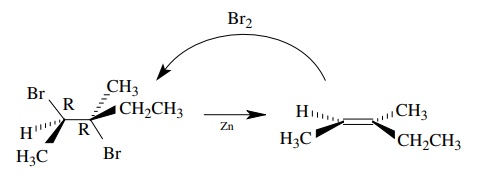
From
the stereochemistry shown, the zinc reduction is a trans elimination of two
bromines. Since the addition of bromine to an olefin is also trans, you could
not use this sequence to isomerize an olefin.
