Olefin Metathesis
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Carbon-Carbon Bond Formation Between Carbon Nucleophiles and Carbon Electrophiles
In the last 10 years or so an exciting new strategy has emerged for the formation of carbon–carbon double bonds, namely olefin metathesis.
OLEFIN METATHESIS
In
the last 10 years or so an exciting new strategy has emerged for the formation
of carbon–carbon double bonds, namely olefin metathesis. This work grew out of
the development of Ziegler–Natta catalysts for the polymerizarion of cyclic
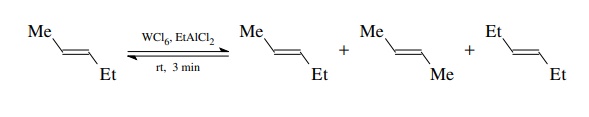
olefins. It was found that when 2-pentene was treated with a catalyst prepared
from tungsten hexachloride and ethylaluminum dichloride, a mixture of
2-pentene, 2-butene, and 3-hexene was produced in minutes at room tempera-ture
(rt)!
It
was shown that the mixture was an equilibrium mixture. Thus it appears that the
alkenes are being broken apart at the double bonds and the pieces reassembled
randomly. This process was termed olefin metathesis because the ends of the
carbon–carbon double bonds are being interchanged.
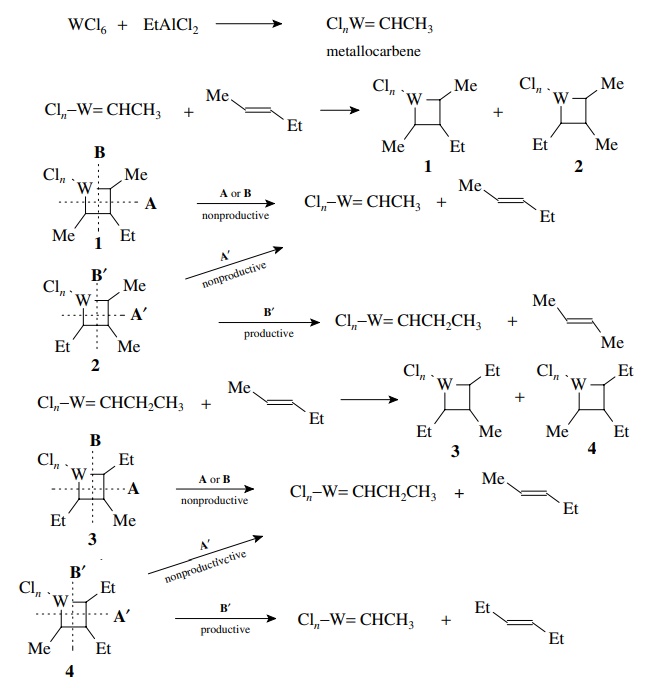
It
was subsequently shown that the active catalytic species is a metallocarbene
complex which contains a carbon–metal double bond. This catalyst undergoes a 2+2 addition with an alkene to give
two metallocyclobutanes 1 and 2 (below) which depend on the regiochemistry
of addition. Metallocyclobutane 1 is
a non-productive intermediate since cleavage across the ring by A or B gives back the reactants. Metallocyclobutane 2 can cleave across the ring A’
to give the original alkene and metallocarbene or it can cleave in the opposite
direction B to give a new alkene and
a new metallocarbene. The new metallocarbene can add the starting material to
give two metallocyclobutanes 3 and 4. It is seen that 3 is a nonproductive intermediate whereas 4 can give another new alkene by cleavage in the B direction. Since each step is
reversible, it can be seen that ultimately the ends of the double bond will be
exchanged to give a mixture of olefins.
Olefin
metathesis is a unique reaction and is only possible by transition metal
catalysis. In fact only complexes of Mo, W, Re, and Ru are known to cat-alyze
olefin metathesis. Once it was known that metallocarbenes were the actual
catalytic species, a variety of metal carbene complexes were prepared and evalu-ated
as catalysts. Two types of catalysts have emerged as the most useful overall.
The molybdenum-based catalysts developed by Schrock and ruthenium-based
catalysts developed by Grubbs.
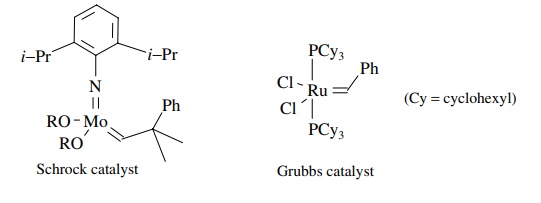
Both
are stable metallocarbene complexes, but they have very different reac-tivity
profiles. The molybdenum catalyst is highly reactive and is effective with
sterically demanding olefins. Its drawbacks are that it is not highly tolerant
of diverse functional groups and has high sensitivity to air, moisture, and
solvent impurities. The ruthenium system, on the other hand, is catalytically
active in the presence of water or air, and it exhibits a remarkable functional
group tolerance. It is not a reactive as the molybdenum catalyst, particularly toward
sterically bulky substrates. However, it is readily available and is the
reagent of choice for all but the most difficult substrates.
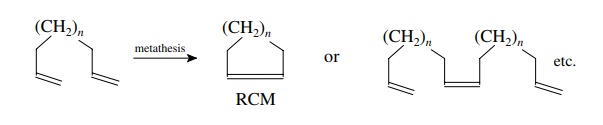
At
present the most synthetically important metathesis process is ring-closing
metathesis (RCM) as it can be used for the preparation of a wide variety of
medium to large ring compounds from acyclic diene precursors. For such dienes
there are two competing processes. The first is RCM, in which the two olefinic
units undergo intramolecular reaction to give a cyclic product. The second is
intermolecular metathesis, which yields open-chain dimers or oligomers.
The
rate of formation of the RCM product is given by a first-order rate law since
the metathesis is intramolecular, rateRCM = k1
[catalyst][diene]. The rate of dimer formation is a second-order rate law
because the reaction is inter-molecular, ratedimer = k2
[catalyst][diene]2. Although these two rate laws are not strictly
comparable because of their different orders, at normal concentrations (0.25–1
M) the difference in rates is mostly due to the difference in the rate
con-stants k1 and k2. Given that the
double-bond energies in five- and six-membered rings are very similar to the
bond energy of an open-chain double bond, the ΔH
±
for the two processes are similar. Thus ΔH± cannot be
responsible for a significant difference in the rates of the two reactions.
The
rate constant k1 for the
RCM process is greater than the dimerization rate constant k2 because of a distinct entropic advantage. In RCM the
two reacting bonds are present in the same molecule and two molecules (the
product and ethylene) are formed from one; thus RCM proceeds with a gain in
entropy. The dimerization process causes loss of translational degrees of
freedom because one molecule is formed from two and thus occurs with a loss of
entropy. As a result the formation of five- and six-membered rings have larger
rate constants and thus proceed at faster rates than dimerization under normal
conditions.
The
use of the Grubbs catalyst is largely restricted to terminal dienes. In these
cases the by-product is ethylene. Since this is an equilibrium process, the
ethylene diffuses out of solution, which helps drive the reaction to
completion. Nevertheless the catalyst is highly tolerant of functional groups
and generally gives good yields.
The
formation of medium rings presents challenges for this and most ring-forming
reactions. There are two major reasons for this. The first is that the molecule
must be able to adopt a reactive conformation in which the two double bonds are
in close proximity for reaction to occur. For larger ring sizes where the
distance between double bonds increases, the population of reactive
conforma-tions decreases so the rate slows as well. The second factor is the
increased ring strain in medium rings which develops at the transition state
and thus raises ΔH± and slows the rate
of the intramolecular process. The rate of the intermolecular dimerization
remains roughly the same. Thus the rate of the intramolecular pro-cess for
medium rings slows dramatically while the dimerization rate remains the same
(for a given concentration).
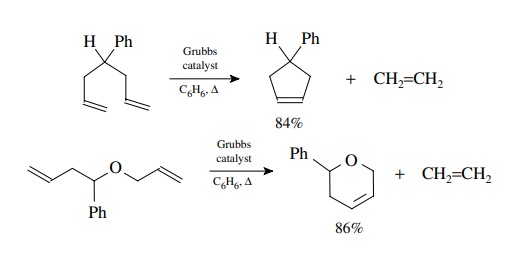
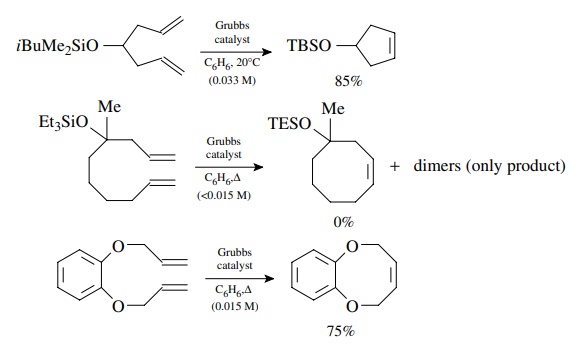
This
behavior is easily demonstrated by the following examples. Formation of a
five-membered ring takes place easily under mild conditions. Formation of a
similar eight-membered ring from an open-chain precursor gave no RCM product but
only dimerization. In contrast bis-allylcatechol gives the eight-membered ring
RCM product efficiently. The phenyl ring provides a conformational constraint
which keeps the olefins in reasonably close proximity and the lack of C–H bonds
in the catechol fragment reduces transannular strain in the eight-membered
ring. These factors allow the RCM to be favored over dimerization.
A
second way to increase the proportion of RCM is to keep the concentration of
the starting diene as low as is feasible. Because dimerization is a
second-order reaction, decreasing the concentration of the diene decreases the
rate of the second-order reaction much faster than the rate of the first-order
RCM reaction decreases. Thus carrying out the reaction at very low
concentrations can actually result in the RCM becoming significantly faster
than dimerization. There are several ways to do this experimentally. The first
is to simply dilute the reaction mixture to the desired concentration and then
run the reaction normally. Although simple, this approach requires a lot of
solvent which must be paid for and then removed. A simpler process is to use
very slow addition of a solution of the diene to a solution of the catalyst. If
the diene reacts as it is added, then the concentration of the diene remains
very low over the course of the addition without having to use large volumes of
solvent to achieve it.
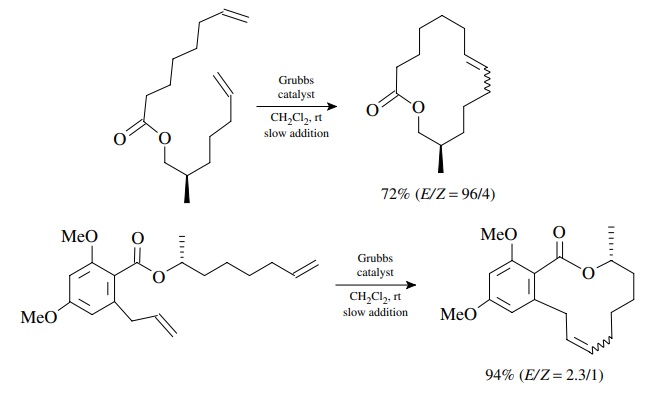
For
large rings where ring strain is again minimal, only the effective
concen-tration of reactive conformers limits the RCM process. In these cases,
running the reaction at high dilution slows the dimerization sufficiently to
allow successful formation of large rings. This is truly a remarkable ability
since large rings are difficult to access by other reactions.
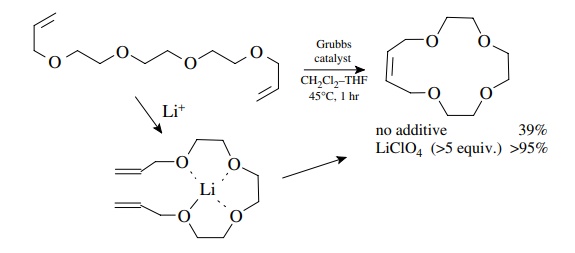
The
effect of conformation on the efficiency of RCM is extremely important. The RCM
of the polyether diene shown below gives only a 39% yield under normal
conditions. Addition of lithium ions to the reaction mixture causes the ether
oxygens to wrap around the lithium cation. This forces the olefinic bonds into
close proximity and the RCM product is now formed in >95% yield. Other structural features such as hydrogen bonding,
restricted rotation (as in amides), or steric effects which cause particular
conformations to be favored are very important in the formation of large ring
systems. Such effects can affect RCM either positively or negatively depending
on whether the olefinic bonds are held proximal or distal to each other.
The
development of RCM has dramatically changed synthetic planning for ring
systems, particularly medium and large ring systems. It represents a new
paradigm for assembling carbon skeletons of ring compounds. Further advances
will only increase its importance as a method for carbon–carbon bond formation.
Related Topics
