Genetic Predisposition to Type B Adverse Drug Reactions
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Mechanisms of Adverse Drug Reactions
TYPE B OR IDIOSYNCRATIC ADVERSE DRUG REACTIONS : Genetic Predisposition to Type B Adverse Drug Reactions
GENETIC PREDISPOSITION TO TYPE B
ADVERSE DRUG REACTIONS
Type
B ADRs have typically been defined to be host-dependent (Rawlins and Thompson,
1991). However, the nature of this host dependency has not been defined for
most drugs, although genetic factors have long been suspected. Indeed, genetic
factors are also important for type A reactions as discussed above. It is
becoming clear that the genetic basis of ADRs, in most cases, is going to be
multi-genic (dependent on a combination of genes) and multi-factorial
(depen-dent on an interaction between genetic and environ-mental factors). This
is going to make it difficult to unravel the genetic basis of adverse reactions
and will require a concerted effort to collect suitable cases and controls as
part of multi-centre international collabo-rations (Pirmohamed and Park,
2001a).
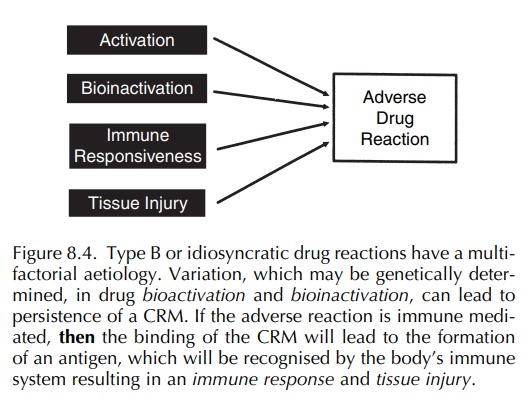
The nature of the polygenic predisposition is unclear but in general could be divided into several areas (Figure 8.4) as follows (Park and Pirmohamed, 2001; Pirmohamed et al., 1998; Pirmohamed and Park, 2001a):
·
Activation: Involves the
activation of drug to CRMs. The
bioactivation of drugs is largely medi-ated by cytochrome P450 enzymes, many of
which have now been shown to be polymorphically expressed (Park, Pirmohamed and
Kitteringham, 1995). Importantly, a deficiency of an enzyme will lead to
reduced bioactivation of a drug and will act as a protective factor. No good
exam-ples have been identified to date. By contrast, the amplification of a
P450 isoform, as seen with
·
CYP2D6 2D6∗2xN (Ingelman-Sundberg, Oscarson and McLellan, 1999), would
increase bioactivation, but again no good example has yet been identified.
·
Detoxification: Absence or reduced
activity of a detoxification enzyme
would lead to a decrease in bioinactivation of the reactive metabolite
(Pirmohamed and Park, 1999) and hence increase the possibility of the reactive
metabolite interacting with important cellular macromolecules result-ing in
different forms of toxicity. The best characterised example of this is the slow
acety-lator phenotype predisposing to hypersensitivity with co-trimoxaole in
HIV-negative patients (Rieder et al.,
1991) and SLE with hydralazine and procainamide (Park, Pirmohamed and
Kitteringham, 1992). There has also been inter-est in the role of the
glutathione-S-transferase genes, many of which have been shown to be
polymorphically expressed. However, although these gene polymorphisms may be
important with respect to certain cancers, studies to date have not shown any
association of the GST gene polymor-phisms with idiosyncratic drug reactions
observed with co-trimoxazole (Pirmohamed et
al., 2000), carbamazepine (CBZ) (Leeder, 1998) and tacrine (De Sousa et al., 1998; Green et al., 1995b).
·
Immune-response
genes:
The process by which the body’s
immune system recognises a drug/drug metabolite as being foreign or antigenic
and thereby mounts an immune response was conceived to be protective, but,
perversely, this may lead to clinical manifestations typical of
hypersensitivity. The genes encoding for immune responsiveness include MHC,
T-cell receptors and co-stimulatory molecules.
·
Tissue-injury genes: The process by
which an immune response is
translated into tissue injury, the nature and extent of which can be
counteracted by repair mechanisms that limit any tissue damage. Typical
candidates include cytokines, chemokines and prostaglandins.
Since
the completion of the human genome project, there have been some striking
findings in the MHC with respect to its role in the genetic predisposition to
drug hypersensitivity. These are illustrated below with reference to two
compounds, abacavir and CBZ. However, it is important to bear in mind two
important issues with reference to the MHC, which means that much more work is
required in this area of the human genome. First, it is the most polymorphic
region of the genome and exhibits a high degree of linkage disequi-librium.
Therefore an association with one polymor-phism does not necessarily mean that
this is a causal association. Second, the MHC has been sequenced and initial
findings suggest that over 60% of the genes in this area are of unknown
function, with only 40% being involved in the immune response (The MHC Sequencing
Consortium, 1999).
Abacavir Hypersensitivity
Abacavir,
an HIV-1 reverse transcriptase inhibitor, causes hypersensitivity,
characterised by skin rash, gastrointestinal and respiratory manifestations, in
about 5% of patients (Hetherington et al.,
2001).
These
reactions can occasionally be fatal, particu-larly on rechallenge. Mallal et al. (2002) found a strong association
between abacavir hypersensitivity and the haplotype comprising HLA-B∗5701, HLA-DR7 and HLA-DQ3 with an odds ratio of over
This association has now been shown in two other cohorts
(Hetherington et al., 2002; Hughes et al., 2004a,b). The same association
however has not been shown in an African American population presumably because
of ethnic differences in linkage disequilib-rium patterns in the MHC (Hughes et al., 2004a). The association with the
MHC in Caucasians is consis-tent with the immune nature of the reaction and the
identification of drug-specific T cells in abacavir hypersensitive patients
(Dodd et al., 2003; Phillips et al., 2005). By contrast, no
association has been found with
polymorphisms in the genes coding for various abacavir-metabolising enzymes
(Hetherington et al., 2002). Mallal et al. (2002) have proposed that in Caucasians genotyping for HLA-B∗5701 should be performed before the prescription of
abacavir, and indeed in their clinic, this has resulted in a reduction in the
incidence of abacavir hypersensi-tivity (Martin et al., 2004). An analysis of the cost effectiveness of prospective
HLA-B∗5701 genotyp-ing before abacavir hypersensitivity based on a
meta-analysis of three cohorts showed that in Caucasians this would be a
cost-effective strategy (Hughes et al.,
2004b).
Carbamazepine Hypersensitivity
Carbamazepine,
a widely used anticonvulsant, causes rashes in up to 10% of patients, and in
occasional cases, this may be the precursor to the develop-ment of a
hypersensitivity syndrome characterised by systemic manifestations such as
fever and eosinophilia (Leeder, 1998; Vittorio and Muglia, 1995). Rarely, CBZ
can induce blistering skin reactions such as SJS and toxic epidermal
necrolysis, two conditions associ-ated with a high fatality rate (Rzany et al., 1999). CBZ hypersensitivity is a
T-cell-mediated disease (Mauri-Hellweg et
al., 1995; Naisbitt et al.,
2003). CBZ is metabolised to CRMs that have been implicated in the pathogenesis
of hypersensitivity (Pirmohamed et al.,
1992). To date, no polymorphisms in the drug-metabolising enzyme gene
polymorphisms have been associated with susceptibility to CBZ hypersensitivity
(Gaedigk, Spielberg and Grant, 1994; Green et
al., 1995a). Analysis of the MHC has led to the find-ing that CBZ
hypersensitivity syndrome, but not mild maculopapular eruptions, is associated
with the haplo-type TNF2-DR3-DQ2 (Pirmohamed et al., 2001). This has also been borne out in more recent studies
in an extensive analysis of the heat shock protein (HSP) locus, which has shown
that severe but not mild CBZ hypersensitivity reactions are associated with
three SNPs in the HSP-70 locus, two in HSP-70-1 and one in HSP-Hom (Alfirevic et al., 2006). These stud-ies suggest
that in Caucasians the causal variant for CBZ hypersensitivity resides on the
ancestral haplo-type 8.1 (Pirmohamed, 2006). In the Han Chinese, however, the
susceptibility locus has been suggested to be different following the finding
of a strong asso-ciation between CBZ-induced SJS and HLA-B∗1502 (Chung et al., 2004).
In
the future, it may be possible to use a comprehen-sive, densely spaced,
genome-wide SNP map that may screen for pharmacogenetically active genes as
whole genome, unbiased searches (Roses, 2000). SNPs are single-base differences
in the DNA sequence, observed between individuals, which occur through-out the
human genome. The International SNP Map Working Group (2001) has published a map
of 1.42 million SNPs throughout the genome, occurring at an average density of
one SNP every 1.9 kb; by the end of 2005, almost 10 million have been
identified, of which 50 000 code for variants that can lead to amino acid
changes. High-density SNP maps derived from this information will provide an
opportunity to perform SNP profiling to identify genetic factors predispos-ing
to ADRs. However, before this can become a reality, the cost of genotyping
needs to come down. Furthermore, given the need to test for multiple mark-ers
simultaneously, an issue that needs to be consid-ered is the sample size and
the level of statistical significance required to prevent the detection of
false-positive associations. A recent study has reported that for testing 100
000 loci in a genome-wide screen will require a 3-fold greater sample size at a
significance level of 2 5 ×
10−7 (Cardon et al., 2000). This does suggest that
for pharmacogenomic detection of rare adverse events, testing in phases I–III
is not likely be practical and will require prospective storage of samples and
evaluation in phase IV when a problem has been identified.
Related Topics
