Antiviral Drugs
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Antibiotics And Synthetic Antimicrobial Agents: Their Properties And Uses
Most of the antibacterial and antifungal agents described earlier in this chapter have little or no activity against viruses because they target structures or enzyme systems that are only found in bacterial and fungal cells.
ANTIVIRAL
DRUGS
Most of the antibacterial and antifungal agents described
earlier in this chapter
have little or no activity
against viruses because
they target structures or enzyme systems
that are
only found in bacterial and fungal cells.
In contrast to other microorganisms, viruses do not possess the enzymes necessary for their own replication. After
entry into the host cell, the virus uses the enzymes already present, or induces the formation of new ones,
in order to synthesize the
individual components of the virus particle which are then assembled and released from the
host cell. Because viruses literally ‘take over’ the machinery of an infected human
cell, there are very few unique
features of viral replication that
can be exploited for the purposes of achieving
selective toxicity, i.e. creating antiviral agents that inhibit or kill the virus without
harming the
human host.
Prior to the identification of the HIV virus in 1983
there was a very
limited range of effective synthetic antiviral agents;
the fourth edition
of this book,
published in 1987, described only nine. The HIV/AIDS pandemic
provided a major stimulus for fundamental research
into the structure and reproduction of viruses in general and retroviruses in particular, and this, together with (1) better understanding of the role
played by some
viruses in the development of specific cancers,
(2) more sophisticated diagnostic methods,
and (3) elucidation of the genomes of several viruses,
has led to a wealth
of new antiviral drugs; Table
11.9 lists 21, and a further 20 that are used
largely or exclusively for the treatment of HIV are shown
in Table 11.10.
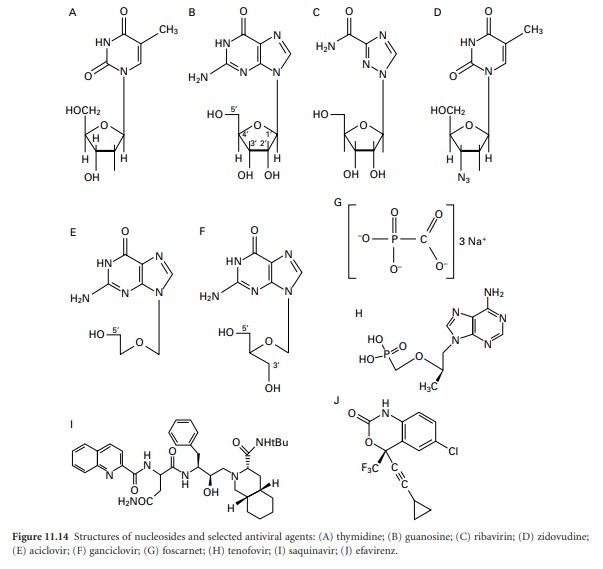
Although these new
drugs may be categorized on the basis of their chemical
structure (Figure 11.14) or mode of action,
most of them are licensed
for use against a limited number
of viruses, and often just
a single one,
so the most convenient and useful way of
considering antivirals is on the basis of the infections they are intended to treat.
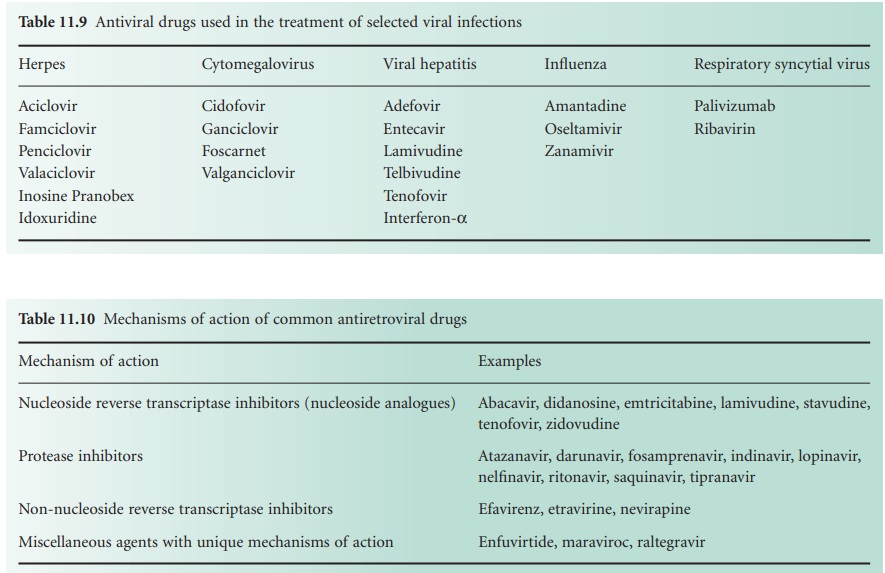
HIV
There is a large and progressively increasing variety of antiretroviral
agents available to treat HIV, and their
use requires specialist knowledge. A detailed account
of the characteristics of each individual drug is beyond the scope of this chapter, but it is possible to gain a good
understanding of the principles of HIV chemotherapy by considering the life cycle
of the virus
(Figure 11.13) in relation
to the modes of action
of the drugs in current use.
The virus particle initially binds to a CD4 protein receptor and one of two co-receptors on the surface
of a T-lymphocyte. Maraviroc,
an antagonist of the CCR5 co-receptor, is licensed in the UK for the treatment of patients exclusively infected with CCR5-tropic HIV. The bound virus then fuses
with the host cell membrane and the viral RNA
is released into
the cell. This
step is targeted by enfuvirtide, a fusion inhibitor, that is used for managing infection that has failed to respond
to a regimen of other anti-retroviral drugs. The single-stranded viral RNA is used as a template
from which a complementary DNA strand is manufactured by viral reverse
transcriptase; there are several
nucleoside analogue inhibitors of this enzyme (Table 11.10),
together with a smaller number
of non-nucleoside
inhibitors (e.g. efavirenz, etravirine, and nevirapine).
The DNA is duplicated, and in its double-stranded form it enters the cell nucleus where an HIV enzyme integrates it into
the host DNA
to create what
is termed a provirus. The integrase enzyme
can be inhibited by raltegravir, a drug which
again is largely
reserved for the treatment of HIV infection
resistant to multiple anti-retrovirals.
The provirus
may remain latent (dormant) within the cell nucleus
for a period of time
varying from 2 weeks
to 20 years; it is then activated by regulatory proteins termed transcription initiators
which cause the viral DNA to be transcribed into viral mRNA and several long protein molecules. It is the latent provirus that represents the major hurdle to complete eradication of HIV, and recent research
interest has focused
on the transcription
initiators as alternative potential targets for
anti-retroviral drug action. The long proteins
only become functional after being split into smaller
molecules by viral protease enzymes
and there are now many protease
inhibitor drugs available
for the treatment of HIV (Table 11.10).
HIV infection cannot be cured,
but strict adherence to a regimen
of anti-retroviral drugs
can substantially extend
survival. However, there
are several problems that may arise during
therapy, of which one of the most significant is the risk of the virus
becoming resistant. This
risk may be minimized by using combinations of three or more
drugs with different mechanisms of action in regimens
that have become known as highly active anti-retroviral therapy (HAART). Treatment is normally initiated
with two nucleoside reverse
transcriptase inhibitors and a non-nucleoside reverse transcriptase inhibitor;
the regimens currently recommended in the UK contain
either tenofovir, emtricitabine and efavirenz, or abacavir, lamivudine, and efavirenz. Retinovir, used in low concentrations at which it has no intrinsic antiviral
activity, has been shown to increase
the duration of effective blood concentrations
of almost all the other protease inhibitors listed in Table 11.10, and these synergistic combinations have given rise to the term boosted protease
inhibitor; a combination product
of retinovir and lopinavir is commercially available. Regimens containing two
nucleoside reverse transcriptase inhibitors and a boosted protease inhibitor are reserved for patients with resistance to firstline treatment. Synergy is often observed between
anti-retroviral drugs, both between agents
having the same, and
different, modes of action.
Other problems of HIV therapy
are drug toxicity
and patient adherence to their prescribed medication, and these
two are often
linked. The variety
and severity of the
side effects, particularly those relating
to redistribution of body
fat, make it more difficult for patients to achieve
the adherence and persistence required
for effective treatment. All the common drugs used in therapy are orally
active because
any drug that required lifelong
daily injections would so predispose to non-adherence as to prejudice its commercial viability. Several of the anti-retroviral drugs listed
in Table 11.10, particularly those with unique mechanisms of action, are used in such restricted circumstances that they
are extremely expensive.
HERPES AND CYTOMEGALOVIRUS INFECTIONS
There are eight herpes viruses capable of causing
human infection, but of these
the most important are:
·
the two herpes simplex viruses, HSV-1 and HSV-2, which, respectively, cause cold sores on the face and lips, and
genital herpes
·
the varicella zoster
virus causing chickenpox and shingles
·
the Epstein–Barr virus responsible for
infectious mononucleosis (glandular fever)
·
the cytomegalovirus (CMV) which may cause
retinitis (inflammation of the retina) and, infrequently, similar symptoms to infectious mononucleosis.
The first
four of the drugs listed
in Table 11.9 for the
treatment of herpes infections are the most important.
Inosine pranobex is an orally
active immuno-modulator the effectiveness of which has not been proven, and idoxuridine,
a pyrimidine analogue
used as a solution in dimethyl-sulphoxide for the treatment of cutaneous herpes, has largely
been superseded by aciclovir.
Many antiviral drugs are nucleoside analogues, and aciclovir together with its prodrug
valaciclovir, and penicyclovir
and its prodrug famciclovir, are important
examples of this group. Aciclovir and peniciclovir are structurally very similar and they act in the same way both
to inhibit viral DNA polymerase and to cause premature termination of DNA synthesis. Both drugs are only effective when they have been phosphorylated in the cell, and selective toxicity
arises because phosphorylation is achieved much more efficiently by virus-encoded thymidine kinase than by the corresponding mammalian enzyme, so human
cell DNA synthesis is little affected.
All four drugs
are used primarily to treat herpes
simplex and varicella zoster
infections; CMV is normally resistant to them and Epstein–Barr virus shows intermediate sensitivity. Aciclovir is available as an intravenous injection, tablets and a cream,
but the last
two of these need to be
administered five times
daily in order
to maintain effective levels. Its prodrug
valaciclovir has a much longer half-life and the dose frequency is correspondingly reduced to 2–3 times
a day. Peniciclovir has little oral activity and is used topically, primarily for cold sores,
but it is applied every
2 hours during
waking hours. The orally active famciclovir is taken between
one and three times daily for the treatment of genital herpes and shingles.
Because CMV does not produce thymidine kinase it is not normally susceptible to aciclovir and
peniciclovir, so a different group of drugs
is used to treat it. Ganciclovir
has a similar structure to aciclovir but it is phosphorylated more effectively in infected cells than healthy ones, albeit by a host cell enzyme.
It is injected intravenously for the treatment of life-threatening or sight-threatening CMV infections in immuno-compromised
patients only, or for the prevention of CMV infection following organ transplants. Valganciclovir is an expensive, orally active,
valine ester prodrug
of ganciclovir which is used
in similar circumstances. Both drugs are toxic, and the latter carries
a specific warning
in the UK British National Formulary about potential
teratogenic and carcinogenic activity. Cidofovir is a cytosine analogue that is active against most
herpesviruses, and is
used by injection in AIDS patients to treat CMV retinitis that is unresponsive to other drugs.
Foscarnet, too, is given for CMV retinitis
when ganciclovir cannot be
used; it has
a relatively simple
phosphonoformic acid structure capable of chelating metal ions,
which is thought to be the basis of
its inhibitory action on polymerase enzymes.
VIRAL HEPATITIS
Hepatitis, inflammation
of the liver, can be caused by various drugs and toxins, but hepatitis due to viral infection is more common. The viruses most
frequently responsible are
the hepatitis viruses
A–E (which are not
all related), but about 5% of cases of viral hepatitis are due to other
viruses, e.g. herpesvirus, CMV, Epstein–Barr virus, etc. Hepatitis virus A (formerly known as infectious hepatitis)
is a self-limiting, rarely fatal,
food-borne infection that
does not result
in permanent liver
damage and is not normally treated
with antiviral drugs.
Hepatitis E is similarly self-limiting and relatively uncommon, and hepatitis D can only
arise as a co-infection with
the hepatitis B virus,
so it is hepatitis viruses
B and C (HBV and
HCV respectively) that are the most problematic and which require antiviral therapy.
It has been estimated that approximately one third
of the world’s
population are infected
with HBV and just
over one tenth
of that number
with HCV. The two infections have several
features in common,
although the viruses
are not of the same family.
In both cases the
disease may be acute or chronic and
in the latter case there may be progression to liver damage
and a higher
risk of contracting liver cancer.
Acute HBV cases do not normally receive antiviral drugs, and
that was formerly
the case for
HCV also, but recent evidence suggests that early treatment of HCV has a
higher success rate and shorter treatment time than that required for chronic
disease, so this practice is now more common.
HBV is unusual in that it is not a retrovirus like HIV but it does use reverse
transcriptase in its replication
process. For this reason two of the drugs used to treat HIV
infection, lamivudine and tenofovir, are also effective for HBV; in addition, adefovirdipivoxil, entecavir and telbivudine are also used,
typically for 6 months or more
for all five drugs. In patients with decompensated liver disease (liver cirrhosis with fluid
accumulation in the abdomen)
lamivudine or adefovir are recommended. Adefovir is effective in lamivudine-resistant HBV infection, but telbivudine should
not be used because cross-resistance may arise.
Entecavir is effective in patients not previously treated with nucleoside
analogues, but resistance can occur
in patients who have received
lamivudine. Again, all of these antivirals are available as relatively
expensive, oral products.
There are several genotypes of HCV which
exhibit different drug sensitivities, so the genotype
should be determined before
drug selection. Interferon-α is used together with ribovirin
for the treatment of chronic infection by both HCV and HBV. Interferons are low molecular weight proteins
produced by virus-infected cells that themselves induce
the formation of a second protein inhibiting the transcription of viral mRNA. Interferon-α needs to be injected
on a daily basis or at
least three times weekly,
but in its polyethylene glycol- derivatized form (peginterferon-α2a) it is much longer
acting and requires less frequent dosing. The combination of ribavirin and interferon-α is less effective against HCV than the combination of peginterferon-α2a and ribavirin. Peginterferon-α2a alone
should be used if ribavirin is contraindicated or not tolerated, but ribavirin alone is ineffective.
INFLUENZA AND RESPIRATORY SYNCYTIAL VIRUS
There are
three related influenza viruses, A, B and C, of
which C is relatively rare and causes
only mild infections. All three reproduce in the same
way and possess
the enzyme
neuraminidase which is responsible for liberating the newly formed virus
particles from the host cell. There are two neuraminidase inhibitors available, oseltamivir which is formulated as an ethyl
ester prodrug for oral administration, and zanamivir which is administered by inhalation. Neither drug has activity
against other viruses.
Both oseltamivir and zanamivir are most effective
if taken within 48 hours of the onset of symptoms, when they may reduce
the duration of the symptoms
by about 1–1.5 days; they
may also reduce
the risk of complications
in elderly
patients or those
with chronic diseases. They are useful too for postexposure prophylaxis of influenza, but again need
to be administered within a few hours
of exposure. Mutant strains
of influenza viruses
have developed resistance to oseltamivir but currently these are
uncommon, and no such resistance has so far been
observed with zanamivir.
Amantadine is a drug
that inhibits an ion-channel protein in influenza A but not influenza B virus (which does not possess
the target molecule). Again, it is effective if given soon after the onset of symptoms, but resistance
has now become widespread so the drug,
although still available,
is no longer recommended in the UK.
Respiratory syncytial
virus (RSV) is a commonly occurring virus related to those causing measles and mumps. It infects most infants by the age of 2 years, but unfortunately there is no long-lasting immunity following infection and no vaccine
available. Ribavirin (Section 13.3) is one of the few antiviral agents
used in the treatment of RSV,
but its value is very much in doubt. Palivizumab
is a monoclonal antibody used for preventing serious lower
respiratory tract disease
caused by RSV in children at high risk of the disease.
Related Topics
