Structural Effects on Acidity
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Acidity and Basicity
Now that a method is in hand to compare acid strengths quantitatively and predict the position of acid–base equilibria.
STRUCTURAL EFFECTS ON ACIDITY
Now
that a method is in hand to compare acid strengths quantitatively and predict
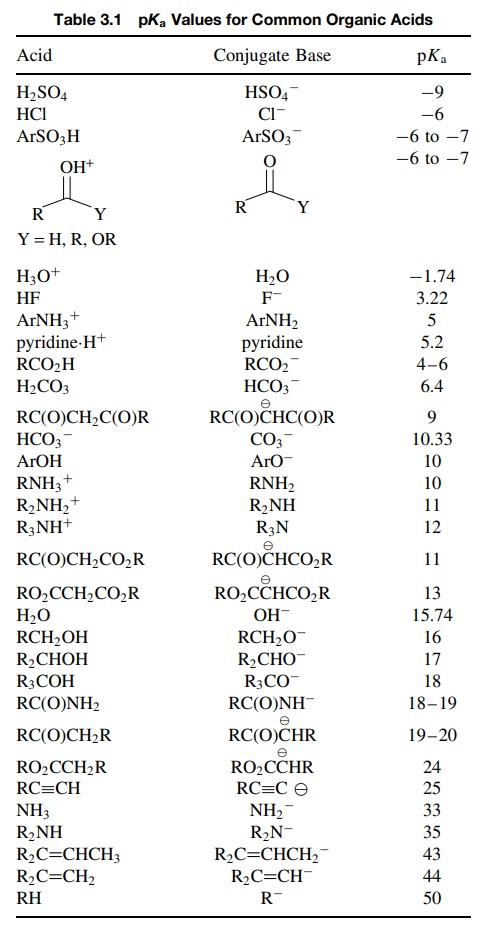
the position of acid–base equilibria, a look at Table 3.1 reveals that organic
compounds have an enormous range of acidities from very strong acids such as
arenesulfonic acids (pKa = −6.5) and protonated carbonyl compounds (pKa = −7 to pKa = −10)
to very weak acids such as alkanes (pKa = 50) and alkenes (pKa =
45). This huge range of acidity of ∼ 1060 is
reflective of the huge diversity of structural elements present in organic
compounds.
How
the structure of an acid influences its pKa
provides a quantitative way to compare the structure of a compound with its
reactivity (in this case acidity). Such structure–reactivity correlations are
crucial for our understanding of how reactions take place and for being able to
predict how a structural change will affect the outcome of a reaction. The
ability to predict how a reaction will respond to changes in structure (or
other variables) takes us out of the realm of trial and error and into the
realm of rational approaches to chemical transformations. Let us examine
briefly some of the structural features which are major influences on acidity.
Returning
to the dissociation of an acid in water, it is seen that the process has some
energy costs and some energy gains. It is this energy balance ( ) which
determines the equilibrium constant and hence the strength of an acid. The
dissociation of an acid in water has an energy cost from the breaking of a bond
to hydrogen and the separation of charges produced by the ionization.
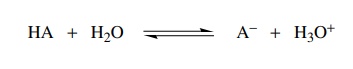
However,
there is an energy gain from the formation of the OH bond of the hydronium ion
and the solvation of the anion by hydrogen bonding to the solvent and solvation
of the hydronium by its hydrogen bonding to the solvent. If a series of different
acids is now compared, it becomes clear that a major energetic difference in
the dissociation of various acids in water is the stability of the conjugate
base and its interaction with the aqueous solvent.
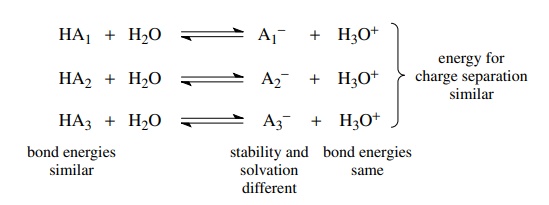
This
is because the other energetic factors which influence the equilibria are
similar for different acids. Bonds from hydrogen to the first-row elements have
similar bond strengths (±5
kcal/mol) so the energy cost of breaking the bond to hydrogen is relatively
constant for most acids of first-row elements. This analysis is especially true
for acids with the proton bonded to the same element. Moreover, since the
solvent is always water, the energy required to separate charges is about the
same. Finally, the H–O bond of the hydronium ion is the same, regardless of
which acid supplies the proton. Consequently, the principal differences in the ΔG’s
of ionization for various acids are due to the differences in stability of their
conjugate bases in the reaction mixture.
This
analysis suggests that structural features which stabilize the conjugate base
(often an anion) will therefore increase the acidity of an acid. While there
are exceptions to this general approach (e.g., comparison of the acidities of
acids in the second and third rows of the periodic table), it provides a sound
basis for predicting what structural factors can increase or decrease the
acidity of organic acids.
There
are three principal factors that lead to increased stability of anions: (a) the
electronegativity of the atom carrying the negative charge, (b) inductive
effects which can stabilize negative charge, and (c) resonance effects which
delo-calize the negative charge over several atoms and hence stabilize the
anion.
Electronegativity
Increased
electronegativity of an atom allows it to carry negative charge more readily,
and the stability of the anion is increased. It is for this reason that the
order of acidity of first-row hydrides is C–H < NH < –OH < FH. Transfer of a proton from these
substances to water yields a series of anions whose stabilities are ordered
according to the electronegativity of the negatively charged atom.
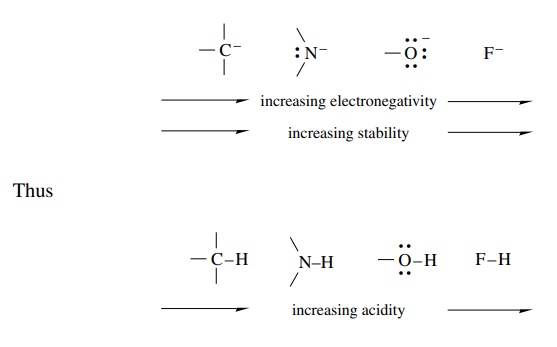
Such
ordering is valid only for elements in the same row in the periodic table.
Comparisons between acids in which the proton is lost from elements from
dif-ferent rows is not valid because the bond strength to the acidic hydrogen
changes greatly from row to row. In the above analysis bond strength is assumed
to be relatively constant; thus it cannot be neglected when significant bond
strength differences between acids are present.
The
effective electronegativity of the atom carrying the charge is also depen-dent
on the hybridization of that atom. As the s character of an orbital increases,
electrons in that orbital are more stable due to greater attraction to the
nucleus. Thus the effective electronegativity of the atom increases. This
effect is clearly seen in the relative acidities of hydrocarbons. Removal of a
proton from alkanes, alkenes, and alkynes produce conjugate bases with electron
pairs in sp3, sp2, and sp orbitals, respectively. As the
amount of s character increases from 25 to 33 to 50% in this series, the
stability of the conjugate base increases and accounts for the marked increase
in acidity in the series. Based on these data, it is expected that
cyclopropane, which because of ring strain has the hydrogens bonded to carbons
which are hybridized at about an sp2.5
level (29% s character), should have a pKa
between that of an alkane and an alkene. In fact, the pKa of cyclopropane is 46 as predicted.

The
increase in acidity by 25 orders of magnitude between sp3- and
sp-hybridized carbon acids is similar to that found for the difference in
acid-ity between an ammonium ion (sp3 hybridization) and a
protonated nitrile (sp hybridization). It is clear that the hybridization of
the orbital they occupy can play a major role in stabilizing electron pairs and
thus influencing the effective electronegativity of an atom.
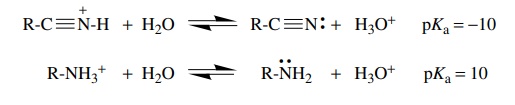
Inductive Effects
The
inductive effect is the ability of a substituent or group near the acidic
proton to alter the electron distribution at the reaction center by
through-bond displace-ment of electrons. The result is that substituents which
withdraw electrons from the reaction center by the inductive effect stabilize
anions and thus increase the acidity of the conjugate acids of those anions.
Conversely, groups which donate electrons make the reaction center more electron
rich and thus make the for-mation of the anion at that center more difficult.
The conjugate acid is thus a weaker acid.
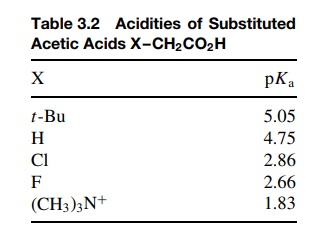
This
is easily demonstrated by considering a group of substituted acetic acids
(Table 3.2). Compared to acetic acid (X = H), replacement of a hydrogen by
more electron-withdrawing groups [Cl, F, (CH3)3N+ ] leads to an
increase in the acidity. Replacement of hydrogen with an electron-donating t -butyl group decreases the acidity. We
can understand these changes in terms of the inductive effect. If we compare
the conjugate bases of acetic acid and chloroacetic acids, it is seen that the
carbon–chlorine bond has a dipole moment associated with it.
This
bond dipole induces smaller dipole moments in adjacent bonds, which in turn
induces ever smaller dipole moments in adjacent bonds.
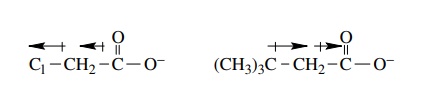
The
result of this inductive effect is that the electron density on the carboxylate
anion is reduced, the negative charge is distributed over more atoms, and the
chloroacetate anion is stabilized relative to acetate. Because the
chloroacetate anion is more stable than the acetate ion, its conjugate acid,
chloroacetic acid, is a stronger acid than the conjugate acid of the acetate
ion, acetic acid (Table 3.1).
As
is expected, groups with higher electronegativity (X = F) or electron defi-ciency result
in greater inductive electron withdrawal, the anion is more stable, and the
acidity is increased. Conversely, a group such as t -butyl is electron donating relative to hydrogen. Its inductive
effect serves to increase the electron density on the carboxylate group,
destabilize the anion, and thus decrease the acidity of its conjugate acid.
As
mentioned, inductive effects operate through bonds by successive bond
polarizations. As such, they diminish rapidly with distance so that very little
effect results if an inductive effect must be transferred through more than
four bonds. As seen in Table 3.3, placement of a chlorine substituent next to
the carboxyl group causes a hundredfold increase in acidity. Moving it to the β position reduces the effect
significantly, while a γ -chloro
substituent causes almost no acidity increase.

Inductive
effects serve to alter the electron distributions in molecules, and
con-sequently they are very important influences on many types of reactions—not
just acidity and basicity. To the extent that electronic changes occur during
the con-version of reactants to products, inductive effects can facilitate or
impede those electronic changes and thus change the rates of conversion. It is
important then to keep them in mind when other examples of reactivity changes
are discussed.
Resonance Effects
A
final structural effect which influences acidity is the delocalization of
electrons via resonance. In terms of acid–base behavior, resonance
delocalization can sta-bilize the conjugate base of an acid, thus making the
acid a stronger acid. For example, alcohols have pKa’s of ∼16 whereas
carboxylic acids have pKa’s
of ∼5. In each case the acidic proton is
lost from oxygen. The bond strength to the proton and the electronegativity of
the atom carrying the charge (oxygen) are identical; thus these factors cannot
account for the large difference in acidity. On the other hand, the alkoxide
ion is a localized anion with the oxygen atom carry-ing a full negative charge
while the carboxylate ion is resonance delocalized. In the carboxylate ion the
electron pair and negative charge are distributed between both oxygens so that
each oxygen carries only a partial negative charge (actually about – 1/2 ) and the anion is greatly
stabilized. Thus carboxylic acids are more acidic than alcohols by ≈1011 or so.
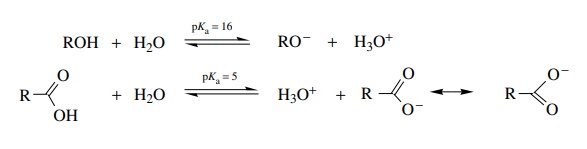
The
following groups of compounds illustrate the profound effect that reso-nance
delocalization has on the stability of anions and hence the acidity of the
conjugate acids. To compare the acidities of these acids, the conjugate bases
can be ranked according to their resonance stabilization and that ranking of
anion stabilization is predictive of the acidity orders.
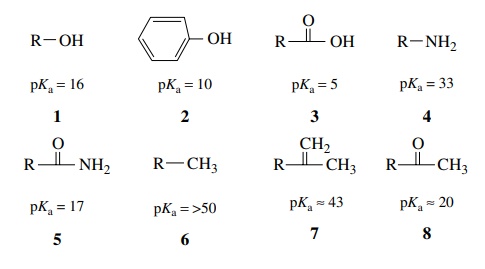
While
resonance stabilization is greatest for those compounds which have more
electron density distributed to more electronegative elements (compare 1, 2,
and 3), delocalization of charge
over any elements results in significant anion stabilization and a corresponding increase in acidity of the
conjugate acid of that anion (e.g., 6,
7, 8). Moreover electronegativity effects can be considered in
addition to resonance effects where applicable. Both amides 5 and methyl ketones 8 have resonance stabilization, but in
amide anions the negative charge is shared between nitrogen and oxygen, while
in ketone enolates the negative charge is shared between carbon and oxygen. Due
to the greater electronegativity of nitrogen over carbon, the amide anion is
more stable and hence amides (pKa
≈ 18–19) are somewhat
more acidic than ketones (pKa
≈ 20–21)
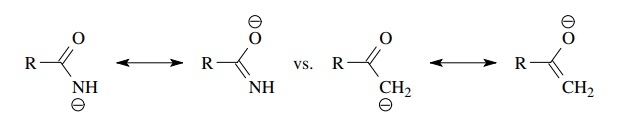
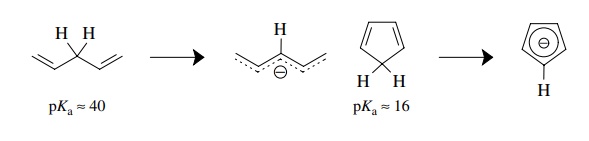
A
particularly strong type of resonance stabilization is found for those
com-pounds which form an aromatic ring upon removal of a proton. The enhanced
aromatic stability of the conjugate base translates into a large increase in
acidity of the acid. Whereas the doubly allylic proton of 1,4-pentadiene is
predicted to have a pKa ≈ 40 due to resonance stabilization
of the anion, the doubly allylic proton in cyclopentadiene has a pKa = 16 because the resulting anion
produces an aromatic π system.
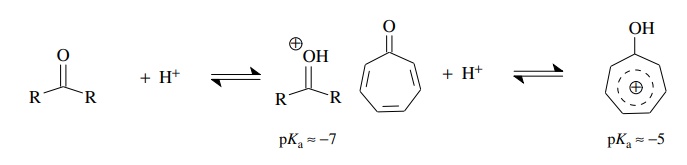
Aromaticity
also explains why tropolone (pKa
≈ −5) is slightly more
basic than a normal ketone (pKa
≈ −7). The conjugate
acid is stabilized upon proto-nation by the formation of an aromatic tropylium
ion.
Inductive
and resonance effects described above can significantly alter the electron
distributions in molecules and can influence not only acidity but many other reactions
as well. A general understanding of these effects will be important in many
different transformations we will encounter.
Related Topics
