Reaction Kinetics
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Mechanisms of Organic Reactions
The first step in delineating the mechanism of a reaction is to determine what reactant species must come together to produce the activated complex of the rate-determining step.
REACTION KINETICS
The
first step in delineating the mechanism of a reaction is to determine what
reactant species must come together to produce the activated complex of the
rate-determining step. This can be done by determining the order of the
reaction with respect to each of the reactants in the process. If the rate of a
reaction is dependent on the concentration of a particular reactant, then that
reactant is involved in the transition state of the rate-determining step of that
reaction. This provides important structural information about the activated
complex since it reveals which chemical species are present in the activated
complex.
The
order of a reaction is found by determining the relationship between rate and
concentration for each reactant. Thus for the elementary process A → B, the rate of reaction ν can be expressed as the decrease in
the concentration of reactant A with time or the increase in the concentration
of product B with time:
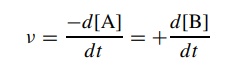
Further,
the rate of the reaction can be expressed as a function of the concen-tration
of A, where k is the rate constant
for the process:
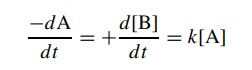
This
differential rate expression shows that the reaction rate is directly dependent
on the concentration of A — the greater is [A], the faster is A converted to B.
The reaction is said to be first order with respect to A because the exponent
of [A] is 1.
The
above expression is the first-order differential rate law for the conver-sion
of A to B. The change in concentration of A over the complete course of the
reaction is given by the integrated rate law, which is found by solving the
differential rate law:
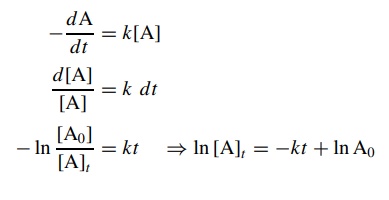
The
integrated rate law shows that the natural logarithm of the concentra-tion of
the starting material A decreases linearly with time. By determining the
concentration of A at various times [At
] and plotting ln[A]t
versus t , a straight line with slope
−k will be obtained if
the reaction is first order in A. (Since the concentration of product at
any time [Bt ] = [A0] − [At ], a plot of the increase in the concentration of B
with time in the form of ln([A0]
− [Bt ]) versus time would also give a straight line whose slope is +k.) If such linear
dependence is observed, then the reaction is first order with respect to A and
the rate constant for the reaction can be determined. If the rate plot is not
linear, then the reaction is not first order with respect to A; that is, a
first-order rate law does not correctly describe the kinetic behavior.
Second-order
reactions occur when two reagents must collide in solution to produce the
activated complex. Thus for the reactions
2A
→ B
A+Β
→ C
each
reaction is second order because the sum of the exponents of species in the
rate law is 2:
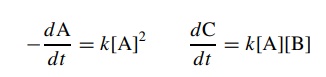
This
means that in the first instance two molecules of A must collide to produce the
activated complex. In the second case a molecule of A and B collide to produce
the activated complex. In each case the second-order dependence requires that both of the colliding molecules are a
part of the activated complex of the rate-determining step.
Integration
of these differential rate laws gives
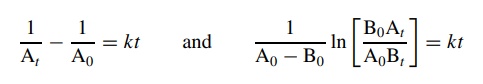
Again
plotting concentration versus time using these integrated second-order rate
laws gives linear plots only if the
reaction is a second-order process. The rate constants can be determined
from the slopes. If the concentration – time plots are not linear, then the
second-order rate equations do not correctly describe the kinetic behavior.
There are integrated rate laws for many different reaction orders.
It
is often possible to simplify the rate law of a second-order process by
employing pseudo-first-order conditions. For a second-order reaction where
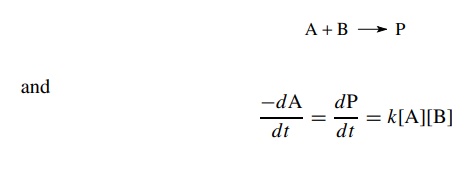
the
integrated rate law for a second-order reaction can be used to plot the kinetic
data. Alternatively, if the concentration of one of the reactants, for example
A, is much larger than the other (10-fold excess minimum, 20-fold better), then
its concentration does not change significantly over the course of the
reaction; thus [A]t ≈ [A]0. The differential
rate law can be approximated as
dP/dt = k[A][B] = k[A0][B] = k’ [B]
where
k = k[A]0.
This new rate law is a first-order equation which is much easier to deal with
and plot. A plot of ln[B] versus t
will give a straight line of slope k’.
From k’ and [A]0 (which is
known), the rate constant can easily be determined. This is a much simpler
method for determining the rate constant than the normal second-order
treatment. The order of the reaction with respect to A can be checked by using
several different initial concentrations of [A]0 and the
relationship
k’= k[A]0
Therefore
log
k’ = log[A]0 + log k
Plots
of log k’ versus log[A]0
will have unit slope if the reaction is first order in A. Alternatively,
doubling [A]0 should double the rate if the reaction is first order
in A.
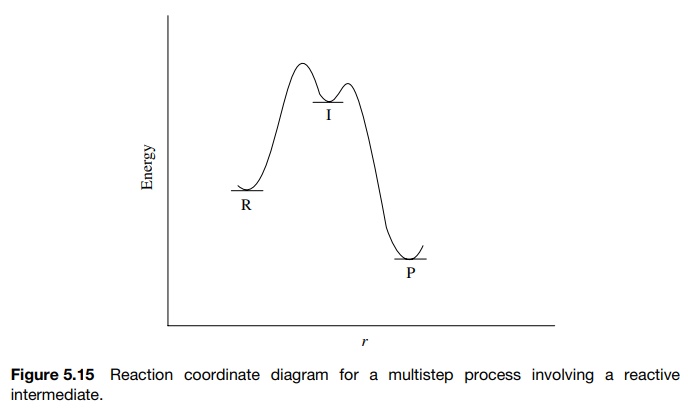
In
a multistep process involving a reactive intermediate, the rate law for the
overall reaction cannot be written down a priori because the step in which the
reactants disappear is different than the step in which the products are formed
(Figure 5.15). In a large number of cases, the intermediate is of high energy
and reacts very rapidly — either returning to reactant or going on to product.
In
such cases the steady-state approximation can be used to derive a rate
expression that can be tested. Thus for a reaction process involving an
intermediate [I]
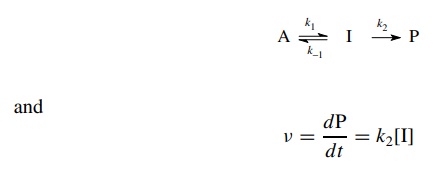
The
concentration of the intermediate [I] which gives product is given by the
difference between the rate of its formation from A and the rate of its
conversion back to reactant A or forward to product P
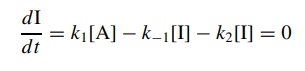
dI/dt = k1[A] − k−1[I] − k2[I] = 0
The
steady-state approximation assumes that since I is very reactive, its
concen-tration will be very low at any time during the reaction and it will not
change appreciably. Therefore dI/d t = 0. Solving the above expression for
the concen-tration of I and substitution into the rate law for the formation
product gives
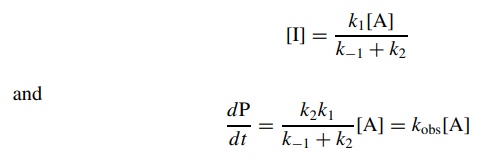
where
the observed rate constant kobs
= k2k1/ k−1 + k2.
This is a first-order rate expression in reactant A and can be integrated and
plotted normally.
Several
limiting cases can be envisioned for such a multistep process. If k2 >> k−1 (the intermediate
goes on to product more rapidly than it returns to reactant),
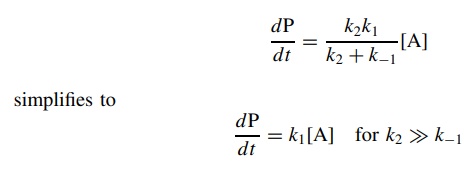
This
represents the case where the first step is rate determining. (If this
situation is known beforehand, one need not work through the steady-state
approximation but merely write down dP/d t = k1A
since the first step is rate limiting and irreversible.)
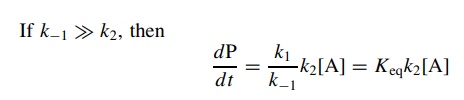
This
represents the case where there is a fast preequilibrium preceding the
rate-determining step. It is again not necessary to work through the
steady-state approximation if this situation is known to exist. If the intermediate
I is in equilibrium with the reactant A, then
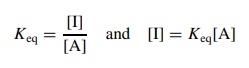
substitution
for [I] in the rate expression gives
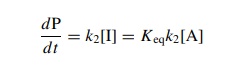
This
is actually the correct way to think about this case since the steady-state
approximation requires that the concentration of I is low, which it may not be
if there is a fast preequilibrium.
If
k2 ≈ k−1, then the full
steady-state rate expression is needed to describe the rate of reaction:
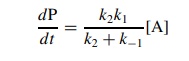
Consider
once again the solvolysis of a tertiary bromide in methanol.
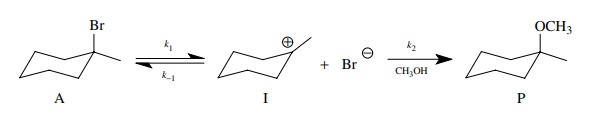
This
reaction can be reduced to the kinetic scheme.
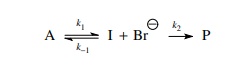
The
rate of product formation is given by the pseudo first-order expression in
which k2 = k[CH3OH]
(normally k2 is taken as
the rate constant because the concentration of methanol is constant),
dP/dt = k2[I]
and
the steady-state approximation is written as
dP/dt = O = k1[A] − k−1[I][Br−] − k2[I] d t
Since
the return of intermediate I to reactants is a second-order reaction, solving
for [I] and substitution into the rate law give
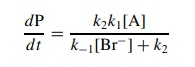
If
k2 >> k−1[Br−], then ν = k1[A]
and simple first-order behavior is found. If k−1[Br] >> k2,
then there is a rapid ionization preequilibrium and
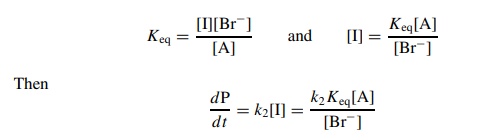
The
rate of product formation will be directly proportional to [A] but inversely
proportional to [Br−].
By using an excess of Br−
so that its initial concentration [Br−]0 does not change
appreciably over the course of the reaction, pseudo-first-order behavior can be
achieved with kobs = k2Keq/[Br−]0.
If
k−1[Br−] ≈ k2,
then the full rate expression will be needed to describe the kinetic behavior.
The rate will be first order in [A] and the rate will slow down in the presence
of added bromide but not in a simple inverse relationship.
Related Topics
