New Drug Development
| Home | | Pharmacology |Chapter: Essential pharmacology : Aspects Of Pharmacotherapy; Clinical Pharmacology And Drug Development
In this era of bewildering new drug introduction and rapid attrition of older drugs, the doctor needs to have an overall idea of the manner in which new drugs are developed and marketed.
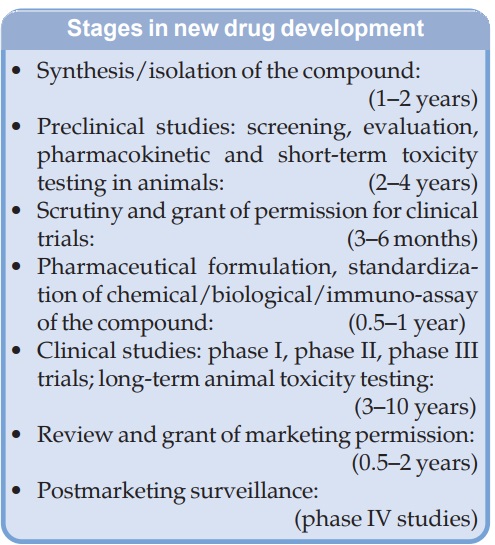
NEW DRUG DEVELOPMENT
In this era of
bewildering new drug introduction and rapid attrition of older drugs, the
doctor needs to have an overall idea of the manner in which new drugs are
developed and marketed. Drug development now is a highly complex, tedious,
competitive, costly and commercially risky process. From the
synthesis/identification of the molecule to marketing, a new drug takes at
least 10 years and costs 500–1000 million US$. The major steps/stages in the
development of a new drug are given in the box.
Approaches To Drug Discovery
Natural Sources
Plants are the oldest source of medicines. Clues about these have been obtained from
traditional systems of medicine prevalent in various parts of the world; Opium
(morphine), Ephedra (ephedrine), Cinchona (quinine), curare
(tubocurarine), belladonna (atropine), Quinghaosu (artemisinin) are the outstanding
examples. Though animal parts have
been used as cures since early times, it was physiological experiments
performed in the 19th and early 20th century that led to introduction of some
animal products into medicine, e.g. adrenaline, thyroxine, insulin, liver
extract, antisera, etc. Few minerals
(iron/calcium salts, etc.) are the other natural medicinal substances. The
discovery of penicillin (1941) opened the floodgates of a vast source— microorganisms—of a new kind of drugs
(antibiotics). The use of microbes
for production of vaccines is older than their use to produce antibiotics.
The above natural sources of medicines are by no means
exhausted, search for new plant, animal and microbial products as drugs is still
a productive approach, especially to serve as lead compounds.
Chemical Synthesis
Synthetic chemistry made its debut in the 19th century and is now the largest
source of medicines. Randomly synthesized compounds can be tested for a variety
of pharmacological activities. Though some useful drugs (barbiturates, chlorpromazine)
have been produced serendipitously by this approach, it has very low
probability of hitting at the right activity in the right compound.
A more practical approach is to synthesize chemical congeners of
natural products/synthetic compounds with known pharmacological activity in the
hope of producing more selective/superior drugs. Many families of clinically
useful drugs have been fathered by a lead compound. Often only ‘mee too’ drugs
are produced, but sometimes breakthroughs are achieved, e.g. thiazide diuretics
from acetazolamide, tricyclic antidepressants from phenothiazines.
Study of several congeners of the lead
compound can delineate molecular features responsible for a particular property.
Application of this structure activity
relationship information has proven useful on many occasions, e.g.
selective β2 agonists (salbutamol)
and β blockers (propranolol,
etc.) have been produced by modifying the structure of isoprenaline, H2
blockers by modifying the side chain of histamine, ethinyl-estradiol by
introducing a substitution that resists metabolic degradation, mesoprostol
(more stable) by esterifying PGE1.
Many drugs are chiral compounds. Because pharmacological activity depends on three
dimensional interaction of drugs with their target biomolecules, the enantiomers (R and S forms or d and l isomers) of chiral drugs differ in biological activity, metabolic
degradation, etc. Often only one of the enantiomers is active. Single
enantiomer drug could be superior to its recemate, because the additional
enantiomer may not only be a ‘silent passenger’ but contribute to side effects,
toxicity (dextrodopa is more toxic than levodopa) load on metabolism or even
antagonize the active enantiomer. Regulatory authorities in many countries, led
by USFDA, have mandated separate investigation of the enantiomers in case the
new drug is a chiral molecule. Approval is withheld unless the pure enantiomers
are shown to be no better than the recemate. Several drugs, originally
introduced as recemates, have now been made available as single enantiomer preparations as well (see box).

Rational Approach
This depends on sound
physiological, biochemical,
pathological knowledge and identification of specific target for drug action such
as H+K+ATPase enzyme or glycoprotein IIa/IIIb receptor. The drug is aimed at
mitigating the derangement caused by the disease, e.g. levodopa was tried in
parkinsonism based on the finding that the condition resulted from deficiency
of dopamine in the striatum. The purine, pyrimidine, folate antimetabolites
were introduced in cancer chemotherapy after elucidation of key role of these
metabolites in cell proliferation. Because virus directed reverse transcriptase
is unique to retroviruses, its inhibitors have been developed as antiHIV drugs.
This approach is very attractive but requires a lot of basic research.
Molecular Modelling
Advances
in protein chemistry and computer aided
elucidation of three dimensional structure of key receptors, enzymes, etc. has
permitted designing of targeted compounds, e.g. designing of selective COX2
inhibitors was prompted by the comparative configuration of COX1 and COX2
enzyme molecules. Study of drug binding to mutated receptors and elucidation of
configuration of drugreceptor complexes is now guiding production of improved
drugs. Attempts are being made to produce individualized drugs according to
pharmacogenomic suitability.
Combinatorial Chemistry
Chemical groups are
combined in a random manner to
yield innumerable compounds and subjected to highthroughput screening on cells, genetically engineered microbes,
receptors, enzymes, etc. in robotically controlled automated assay systems.
Computerized analysis is used to identify putative drugs which are then
subjected to conventional tests. This new approach has vast potentials, but has
not achieved major breakthroughs so far.
Biotechnology
Several drugs are now
being produced by recombinant DNA
technology, e.g. human growth hormone, human insulin, interferon, etc. Some
monoclonal and chimeral antibodies have been introduced as drugs.
New
molecules, especially antibiotics, regulatory peptides, growth factors,
cytokines, etc. produced by biotechnological methods can be evaluated as
putative drugs. Other experimental approaches in new drug development are
antisense oligonucleotides and gene therapy.
Preclinical Studies
After
synthesizing/identifying a prospective compound/series of compounds, it is
tested on animals to expose the whole pharmacological profile. Experiments are
generally performed on a rodent (mouse, rat, guinea pig, hamster, rabbit) and
then on a larger animal (cat, dog, monkey). As the evaluation progresses
unfavourable compounds get rejected at each step, so that only a few out of
thousands reach the stage when administration to man is considered.
Toxicity tests
The aim is to
determine safety of the compound in at least 2 animal species, mostly mouse/rat
and dog by oral and parenteral routes.
Acute toxicity: Single escalating doses are given to small groups of animals that are observed for
overt effects and mortality for 1–3 days. The dose which kills 50% animals (LD50)
is calculated. Organ toxicity is examined by histopathology on all animals.
Subacute toxicity: Repeated doses are given for 2–12 weeks depending on the duration of intended
treatment in man. Doses are selected on the basis of ED50 and LD50.
Animals are examined for overt effects, food intake, body weight, haematology,
etc. and organ toxicity.
Chronic toxicity: The drug is given for
6–12 months and effects are studied
as in subacute toxicity. This is generally undertaken concurrently with early
clinical trials.
Special longterm toxicity: These tests are
generally performed only on drugs which
cross phase I clinical trials.
Reproduction and teratogenicity: Effects on spermatogenesis, ovulation, fertility and developing
foetus are studied.
Mutagenicity: Ability of the drug to
induce genetic damage is assessed in
bacteria (Ames test), mammalian cell cultures and in intact rodents.
Carcinogenicity: Drug is given for longterm, even the whole life of the animal and they are watched
for development of tumours.
Standardized procedures
under ‘Good Laboratory Practices’ (GLP) have been laid down for
the conduct of animal experiments,
especially toxicity testing.
Clinical Trials
When a compound
deserving trial in man is identified by animal studies, the regulatory
authorities are approached who on satisfaction issue an ‘investigational new
drug’ (IND) licence. The drug is formulated into a suitable dosage form and
clinical trials are conducted in a logical phased manner. To minimize any risk,
initially few subjects receive the drug under close supervision. Later, larger
numbers are treated with only relevant monitoring. Standards for the design,
ethics, conduct, monitoring, auditing, recording and analyzing data and reporting
of clinical trials have been laid down in the form of ‘Good Clinical Practice’ (GCP) guidelines by an International Conference on Harmonization (ICH). Adherence to
these provides assurance that the data and reported results are credible and accurate,
and that the rights, integrity and confidentiality of trial subjects are
protected. The clinical studies are conventionally divided into 4 phases.
Phase I: Human Pharmacology And Safety
The
first human administration of the drug is carried out by qualified clinical
pharmacologists/ trained physicians in a setting where all vital functions are
monitored and emergency/ resuscitative facilities are available. Subjects
(mostly healthy volunteers, sometimes patients) are exposed to the drug one by
one (total 20–40 subjects), starting with the lowest estimated dose and
increasing stepwise to achieve the effective dose. The emphasis is on safety
and tolerability, while the purpose is to observe the pharmacodynamic effects in
man, and to characterize absorption, distribution, metabolism and excretion. No
blinding is done: the study is open label.
Phase II: Therapeutic Exploration And Dose Ranging
This is conducted by
physicians who are trained as clinical investigators on 100–400 patients selected
according to specific inclusion and exclusion criteria. The primary aim is
establishment of therapeutic efficacy, dose range and ceiling effect in a controlled
setting. Tolerability and pharmacokinetics are studied as extension of phase I.
The study may be blinded or open label and is generally carried out at 2–4
centres.
Phase III: Therapeutic Confirmation/Comparison
Generally these are
randomized double blind comparative trials conducted on a larger patient
population (500–3000) by several physicians at many centres. The aim is to
establish the value of the drug in relation to existing therapy. Safety,
tolerability and possible drug interactions are assessed on a wider scale,
while additional pharmacokinetic data may be obtained. Indications are
finalized and guidelines for therapeutic use are formulated. A ‘new drug
application’ (NDA) is submitted to the licencing authority, who if convinced give
marketing permission.
Phase IV: Postmarketing Surveillance/Studies
After the drug has
been marketed for general use, practicing physicians are identified through
whom data are collected on a structured proforma about the efficacy,
acceptability and adverse effects of the drug (similar to prescription event
monitoring). Patients treated in the normal course form the study population:
numbers therefore are much larger. Uncommon/idiosyncratic adverse effects, or
those that occur only after longterm use and unsuspected drug interactions are
detected at this stage. Patterns of drug utilization and additional indications
may emerge from the surveillance data.
Further therapeutic
trials involving special groups like children, elderly, pregnant/lactating women,
patients with renal/hepatic disease, etc. (which are generally excluded during
clinical trials) may be undertaken at this stage. Modified release dosage
forms, additional routes of administration, fixed dose drug combinations, etc.
may be explored.
As such, many drugs
continue their development even after marketing.
Related Topics
