Mechanism of Drug Action
| Home | | Pharmacology |Chapter: Essential pharmacology : Pharmacodynamics Mechanism Of Drug Action; Receptor Pharmacology
Only a handful of drugs act by virtue of their simple physical or chemical property; examples are:
MECHANISM OF DRUG ACTION
Only a handful of drugs act by virtue of their
simple physical or chemical property; examples are:
§ Bulk laxatives
(ispaghula)—physical mass
§ Dimethicone, petroleum
jelly—physical form, opacity
§ Paraamino benzoic
acid—absorption of UV rays
§ Activated charcoal—adsorptive
property
§ Mannitol, mag.
sulfate—osmotic activity
§ 131I and other
radioisotopes—radioactivity
§ Antacids—neutralization
of gastric HCl
§ Pot.
permanganate—oxidizing property
§ Chelating agents (EDTA,
dimercaprol)—chelation of heavy metals.
§ Cholestyramine—sequestration
of cholesterol in the gut
§ Mesna—Scavenging of
vasicotoxic reactive metabolites of cyclophosphamide
Majority of drugs
produce their effects by interacting with a discrete target biomolecule, which
usually is a protein. Such mechanism confers selectivity of action to the drug.
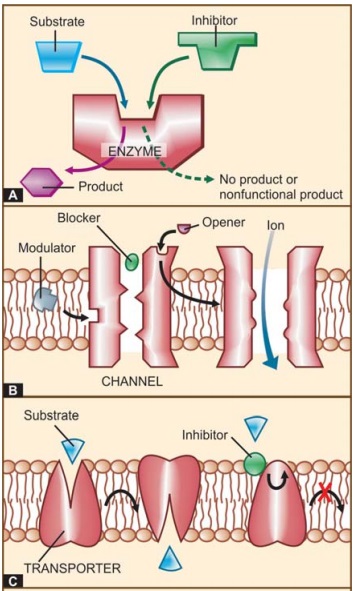
Functional proteins that are targets of drug action can be grouped into four major categories, viz. enzymes, ion channels, transporters
and receptors (See Fig. 4.1).
However, a few drugs do act on other proteins (e.g. colchicine, vinca
alkaloids, taxanes bind to the structural protein tubulin) or on nucleic acids
(alkylating agents).
I. ENZYMES
Almost all biological
reactions are carried out under catalytic influence of enzymes; hence, enzymes
are a very important target of drug action. Drugs can either increase or
decrease the rate of enzymatically mediated reactions. However, in
physiological systems enzyme activities are often optimally set. Thus, stimulation
of enzymes by drugs, that are truly foreign substances, is unusual. Enzyme
stimulation is relevant to some natural metabolites only, e.g. pyridoxine acts
as a cofactor and increases decarboxylase activity. Several enzymes are
stimulated through receptors and second messengers, e.g. adrenaline stimulates
hepatic glycogen phosphorylase through β receptors and cyclic
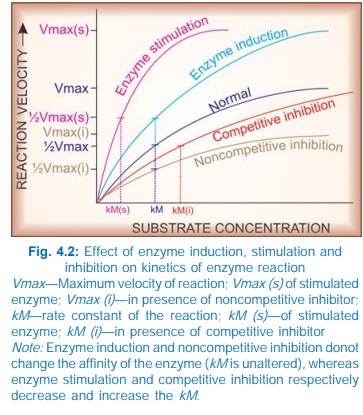
AMP. Stimulation of an enzyme increases its affinity for the substrate so that
rate constant (kM) of the reaction is
lowered (Fig. 4.2).
Apparent increase in
enzyme activity can also occur by enzyme
induction, i.e. synthesis of more enzyme protein. This cannot be called
stimulation because the kM does not
change. Many drugs induce microsomal enzymes.
Inhibition
of enzymes is a common mode of drug action.
A.
Nonspecific inhibition
Many
chemicals and drugs are capable
of denaturing proteins. They alter the tertiary structure of any enzyme with
which they come in contact and thus inhibit it. Heavy metal salts, strong acids
and alkalies, alcohol, formaldehyde, phenol inhibit enzymes nonspecifically.
Such inhibitors are too damaging to be used systemically.
B.
Specific inhibition
Many
drugs inhibit a particular enzyme
without affecting others. Such inhibition is either competitive or noncompetitive.
i) Competitive (equilibrium type) The drug being structurally similar competes with the
normal substrate for the catalytic binding site of the enzyme so that the
product is not formed or a nonfunctional product is formed (Fig. 4.1A), and a
new equilibrium is achieved in the presence of the drug. Such inhibitors
increase the kM but the Vmax remains unchanged (Fig. 4.2), i.e.
higher concentration of the substrate
is required to achieve ½ maximal reaction velocity, but if substrate
concentration is sufficiently increased, it can displace the inhibitor and the
same maximal reaction velocity can be attained.
§ Physostigmine and
neostigmine compete with acetylcholine for cholinesterase.
§ Sulfonamides compete
with PABA for bacterial folate synthetase.
§ Moclobemide competes
with catecholamines for monoamine oxidaseA (MAOA).
§ Captopril competes
with angiotensin 1 for angiotensin converting enzyme (ACE).
§ Finasteride competes
with testosterone for 5αreductase
§ Letrozole competes
with androstenedione and testosterone for the aromatase enzyme.
§ Allopurinol competes
with hypoxanthine for xanthine oxidase; is itself oxidized to alloxanthine (a
non competitive inhibitor).
§ Carbidopa and
methyldopa compete with levodopa for dopa decarboxylase.
A nonequilibrium type of enzyme inhibition
can also occur with drugs which react with the same catalytic site of the
enzyme but either form strong covalent bonds or have such high affinity for the
enzyme that the normal substrate is not able to displace the inhibitor, e.g.
§ Organophosphates react
covalently with the esteretic site of the enzyme cholinesterase.
§ Methotrexate has
50,000 times higher affinity for dihydrofolate reductase than the normal
substrate DHFA.
In these situations, kM is increased and Vmax is reduced.
ii) Noncompetitive The inhibitor reacts with an adjacent site and not with the catalytic
site, but alters the enzyme in such a way that it loses its catalytic property.
Thus, kM is unchanged but Vmax is reduced. Examples are given in
the box.
N
Acetazolamide — Carbonic anhydrase
Aspirin, indomethacin — Cyclooxygenase
Disulfiram — Aldehyde dehydrogenase
Omeprazole — H+ K+ ATPase
Digoxin — Na+ K+ ATPase
Theophylline — Phosphodiesterase
Propylthiouracil — Peroxidase in thyroid
Lovastatin — HMGCoA reductase
Sildenafil — Phosphodiesterase5
II. ION CHANNELS
Proteins which act as
ion selective channels participate in transmembrane signaling and regulate
intracellular ionic composition. This makes them a common target of drug action
(Fig. 4.1B). Drugs can affect ion channels either through specific receptors
(ligand gated ion channels, Gprotein operated ion channels, see Fig. 4.4 and p. 48), or by directly
binding to the channel and affecting ion movement through it, e.g. local
anaesthetics which physically obstruct voltage sensitive Na+ channels (See Ch 26). In addition, certain drugs
modulate opening and closing of the channels, e.g.:
§ Quinidine blocks
myocardial Na+ channels.
§ Dofetilide and
amiodarone block myocardial delayed rectifier K+ channel.
§ Nifedipine blocks Ltype
of voltage sensitive Ca2+ channel.
§ Nicorandil opens ATPsensitive
K+ channels.
§ Sulfonylurea
hypoglycaemics inhibit pancreatic ATPsensitive K+ channels.
§ Amiloride inhibits
renal epithelial Na+ channels.
§ Phenytoin modulates
(prolongs the inactivated state of) voltage sensitive neuronal Na+ channel.
§ Ethosuximide inhibits
Ttype of Ca2+ channels in thalamic neurones
III. TRANSPORTERS
Several substrates are
translocated across membranes by binding to specific transporters (carriers)
which either facilitate diffusion in the direction of the concentration
gradient or pump the metabolite/ion against the concentration gradient using
metabolic energy (see p. 13–15; Fig.
2.5). Many drugs produce their action by directly interacting with the solute
carrier (SLC) class of transporter proteins to inhibit the ongoing
physiological transport of the metabolite/ion (Fig. 4.1C).
Examples are:
§ Desipramine and
cocaine block neuronal reuptake of noradrenaline by interacting with
norepinephrine transporter (NET).
§ Fluoxetine (and other
SSRIs) inhibit neuronal reuptake of 5HT by interacting with serotonin
transporter (SERT).
§ Amphetamines
selectively block dopamine reuptake in brain neurons by dopamine transporter
(DAT).
§ Reserpine blocks the
granular reuptake of noradrenaline and 5HT by the vesicular amine transporter.
§ Hemicholinium blocks
choline uptake into cholinergic neurones and depletes acetylcholine.
§ The anticonvulsant
tiagabine acts by inhibiting reuptake of GABA into brain neurones by GABA
transporter GAT 1.
§ Furosemide inhibits
the Na+K+2Cl¯ cotransporter in the ascending limb of loop of Henle.
§ Hydrochlorothiazide
inhibits the Na+Cl¯ symporter in the early distal tubule.
§ Probenecid inhibits
active transport of organic acids (uric acid, penicillin) in renal tubules by
interacting with organic anion transporter (OAT).
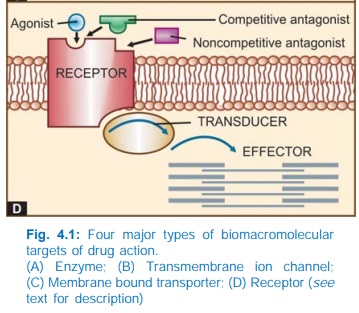
IV. RECEPTORS
The
largest number of drugs do not bind directly to the effectors, viz. enzymes, channels, transporters,
structural proteins, template biomolecules, etc. but act through specific
regulatory macromolecules which control the above listed effectors. These
regulatory macromolecules or the sites on them which bind and interact with the
drug are called ‘receptors’.
Receptor:
It
is defined as a macromolecule or binding site located
on the surface or inside the effector cell that serves to recognize the signal
molecule/drug and initiate the response to it, but itself has no other
function.
Though, in a broad
sense all types of target biomolecules,
including the effectors (enzymes, channels, transporters, etc.) with which a
drug can bind to produce its action have been denoted as ‘receptors’ by some
authors, such designation tends to steal the specific meaning of this important
term. If so applied, xanthine oxidase would be the ‘receptor’ for allopurinol,
Ltype Ca2+ channel would be the ‘receptor’ for nifedipine, serotonin
transporter (SERT) would be the ‘receptor’ for fluoxetine; a connotation not in
consonence with the general understanding of the term. It is therefore better
to reserve the term ‘receptor’ for purely regulatory macromolecules which
combine with and mediate the action of signal molecules including drugs.
The following terms are used in describing drugreceptor
interaction:
Agonist
An agent which
activates a receptor to produce an effect
similar to that of the physiological signal molecule.
Inverse Agonist
An agent which
activates a receptor to produce an
effect in the opposite direction to that of the agonist.
Antagonist
An
agent which prevents the action of an agonist on a
receptor or the subsequent response, but does not have any effect of its own.
Partial agonist
An
agent which activates a receptor to produce
submaximal effect but antagonizes the action of a full agonist.
Ligand
(Latin: ligare—to bind) Any molecule which attaches selectively to particular
receptors or sites. The term only indicates affinity or binding without regard
to functional change: agonists and competitive antagonists are both ligands of
the same receptor.
The
overall scheme of drug action through receptors is depicted in Fig. 4.1D.
