Chemical Shift
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Structure Determination of Organic Compounds
The range of frequencies over which protons absorb in most organic molecules depends on the applied field.
CHEMICAL SHIFT
The
range of frequencies over which protons absorb in most organic molecules
depends on the applied field. For example, for an applied field of 14,000 G,
most protons will absorb over a range of 600 Hz beginning at the value of 60 × 106 Hz (60 MHz), or from
59,999,400 to 60,000,000 Hz. At 23,486 G this range is 1000 Hz near the value
of 100 MHz or from 99,999,000 Hz to 100,000,000 Hz. Thus the actual range of
frequency of absorption depends on the magnetic field of the instrument. (This
is exactly as expected since the energy gap between the spin states and hence
the frequency of absorption are dependent on the applied field.) To compare
absorption values from different instruments, a dimensionless scale must be
devised that is independent of the magnetic field of the instrument. This is
accomplished by using the absorption of tetramethylsilane (TMS) as a spectral
anchor. The frequency of absorption of a given set of protons is measured
relative to the frequency of absorption of TMS. This absorption frequency
difference Δv in hertz (cps) is
expressed as δ, the chemical shift of
the protons in ppm, where
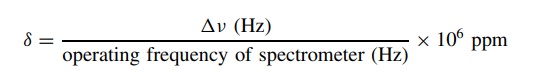
The
chemical shift δ is dimensionless and
independent of the spectrometer. Since normal absorption ranges Δv are about 0 – 600 Hz for an operating
frequency of 60 × 10− 6 Hz, or
0 – 1000 Hz at 100 ×
106 Hz, and so on, chemical shifts range from 0 to 10 ppm for most
protons.
In practice a small amount of TMS (<1%) is added to the NMR sample, the TMS signal is set at 0 ppm, and the protons of the sample are then measured in parts per million relative to TMS. The choice of TMS as a standard is useful because nearly all other protons absorb at frequencies lower than TMS. It is rou-tine to present NMR spectra with low frequency on the left and high frequency on the right (Figure 11.5). Thus the TMS signal defines δ = 0 ppm on the right side of the spectrum and other proton signals are found to the left or downfield from TMS from 0 to about 10 ppm.
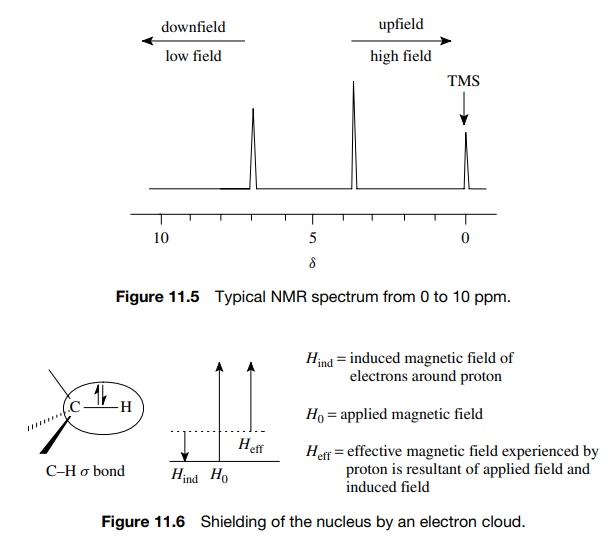
It is also normal to describe signals having larger
chemical shifts as being downfield from protons with smaller chemical shifts.
The left side of the spectrum is termed low
field and the right side high field.
With
a method available to measure differences in chemical shifts between protons,
it is appropriate to ask why different protons experience different Heff’s even though a single H0 is applied to the sample.
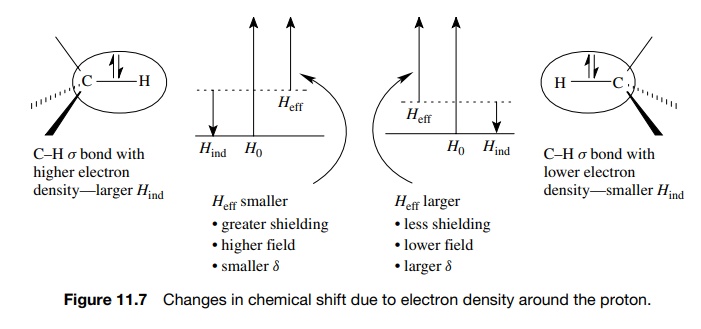
The explanation lies in the fact that nuclei are surrounded by electron clouds
(Figure 11.6). In the applied field H0,
electron pairs in bonds surrounding the hydrogens act to counter the applied
field by induced fields (Hind).
The result is that the nucleus is shielded from the applied field by its
electron cloud. (Nuclei which are more shielded come at higher fields and have
lower chemical shifts.)
Thus
it is the electron density around the nucleus which shields the nucleus from
the applied field. It follows that the greater the electron density around a
proton, the larger will be the induced field Hind and that proton will be more shielded. It will
appear more upfield and will have a smaller chemical shift (δ value). Conversely the lower the
electron density around a proton, the less shielded it will be, the more
downfield it will be, and it will have a larger δ value (Figure 11.7).
Structural
features which withdraw electrons from protons cause downfield shifts and
larger δ values, while structural
features which increase electron density around protons cause upfield shifts
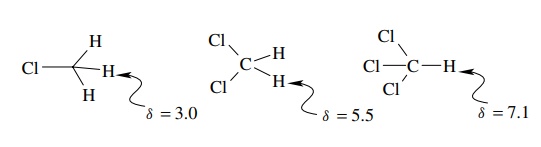
and lower δ values. For example,
chemical shifts for methyl chloride, dichloromethane, and chloroform are δ = 3.0, δ = 5.5, and δ = 7.1, respectively. The inductive effects of increasing numbers of
chlorine atoms decrease the electron density about the hydrogens and result in
increasing chemical shifts.
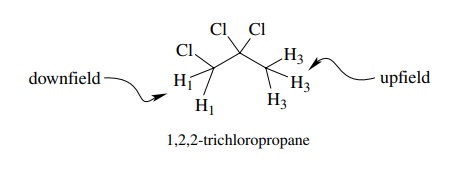
Likewise
1,2,2-trichloropropane discussed previously has the two-proton sig-nal
downfield from the three-proton signal. This is because the methylene protons
are influenced by the inductive effects of three chlorine atoms, two vicinal
and one geminal, while the methyl group is influenced by only two vicinal
chlorine atoms. The electron density is higher at the methyl hydrogens, which
are more shielded and occur at higher fields than the two protons of the
methylene group.
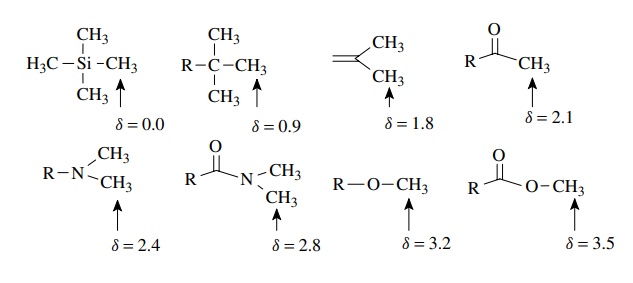
Consideration
of a series of compounds containing methyl groups illustrates clearly the
influence of the electron density on chemical shift. As the
electron-withdrawing ability of groups attached to the methyl group increase,
progressive downfield shifts are evident and δ values increase. Conversely TMS comes very far upfield because
silicon – carbon bonds are polarized toward carbon and result in very high
electron density about the methyl hydrogens of TMS.
Although
the influence of electron density on chemical shift is clear, it is not the
only factor which determines the chemical shift, as seen from the following
series of compounds:
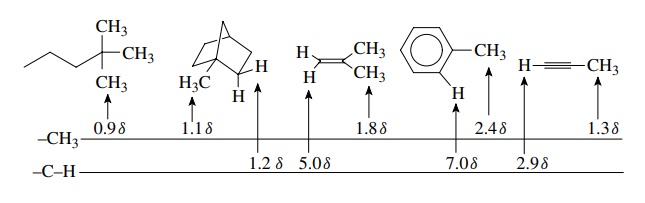
Comparing
the methyl groups, we find that typical saturated aliphatic methyl groups come
at 0.9 – 1.1 ppm. However, attaching a methyl group to a double bond gives a
change to 1.8δ. Attaching the methyl
group to an aromatic ring moves it further downfield to 2.4δ. Attachment to a triple bond moves it back upfield to 1.3δ. Analogous but even larger changes in
chemical shift are seen for protons directly attached to double bonds, aromatic
rings, and triple bonds. Simple electron density shielding arguments cannot
satisfactorily account for these large changes in chemical shift.
For
example, the greater s character of sp2 orbitals and hence greater
effective electronegativity of sp2-hybridized carbon might account
for the downfield shift of the protons of a methyl group when it is attached to
an olefinic carbon rather than a saturated sp3 carbon; however, the
sp2 carbons of aromatic rings should induce the same downfield
shift. In fact, aromatic methyl groups are shifted significantly further
downfield. By the same argument, attachment of a methyl group to the
sp-hybridized carbon of an acetylene, which has even greater s character,
should cause the chemical shift to move even further downfield. In fact,
propargylic methyl groups are found at higher field than allylic methyl groups.
It
is clear that there are other factors at work which influence the chemical
shifts of different types of protons.
Simple
shielding of the hydrogen nucleus by its surrounding cloud of electrons is isotropic in that the induced magnetic
field is the same for any orientation of the hydrogen relative to the magnetic
field. This is due to the fact that the electron cloud around the hydrogen
nucleus behaves as though it is spherical (or nearly so). Other types of
electron clouds (double bonds, aromatic clouds, triple bonds) are not
spherically symmetric. As a consequence, the induced fields for these types of
bonds are not the same at different orientations of the functional group in the
magnetic field. This anisotropic
shielding, or anisotropy, leads
to regions of shielding and deshielding around the functional group that are
averages of the orientations possible.
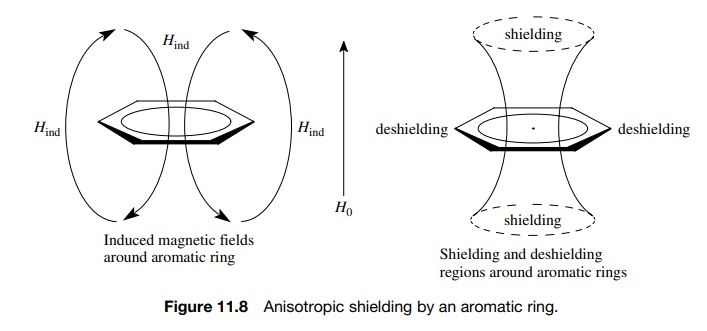
Aromatic
rings have among the strongest anisotropy of any group. Above and below the
ring there is a strong shielding region (Hind
is in opposition to the applied field) while in the plane of the ring there is
a strong deshielding region (Hind
is in the same direction as the applied field). This phenomenon is termed ring current and has been used as a
criterion to establish whether a compound is
aromatic (Figure 11.8). Consequently protons and groups attached to the
ring are in the plane of the ring and thus are strongly deshielded and come at
low fields relative to a comparable proton in a nonaromatic compound. Aromatic
protons normally come at δ > 7 ppm
and benzylic methyl groups come at δ ≈ 2.4, which are both significantly shifted downfield due to the
anisotropy of the aromatic ring. (The shift of benzylic protons is less than
the shift of aromatic protons because they are further from the aromatic ring
than the protons directly attached to the ring.)
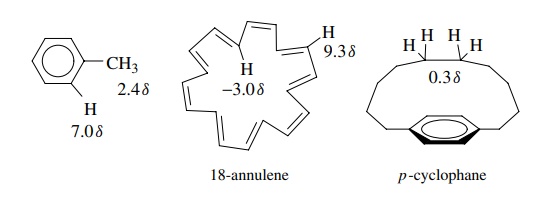
If
protons could be positioned in the center of or above the aromatic ring, they
would fall in the shielding region and should come at high field. For example,
18-annulene is an aromatic compound (4n
+ 2, n = 4). The protons on the outside of
the ring lie in the deshielding region and have δ = 9.3 ppm while those on the inside of the
ring fall in the shielding region and have δ
= −3.0. They come at higher field than TMS
due the anisotropic shielding from the ring current. For the same reason, the
central protons in p-cyclophanes come
at higher fields because they are placed over the aromatic ring in the
shielding region.
Double bonds contain one σ bond and one π bond, which results in anisotropic shielding, as shown in Figure 11.9. There is a conical shielding region normal to the molecular plane and a deshielding region in the molecular plane. This is true for all double-bonded functional groups such as olefins, carbonyl groups, and imines, and it explains why olefinic protons (δ ≈ 5) and aldehyde protons (δ = 9 – 10) absorb at such low fields.
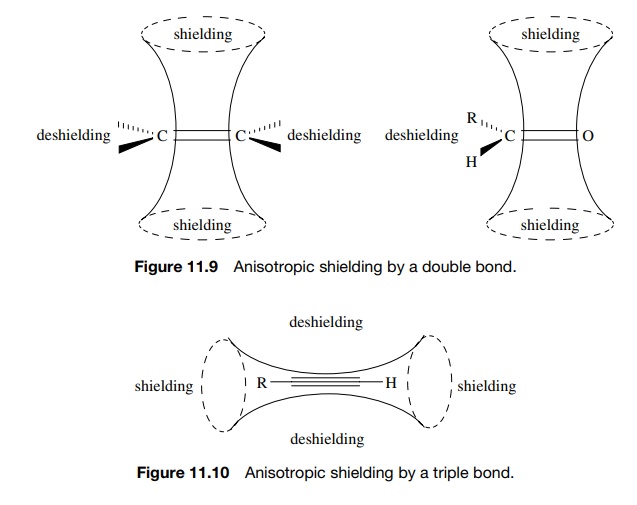
Acetylene
(and nitriles), because of their cylindrical symmetry, have shielding regions
along the triple-bond axis (Figure 11.10). Thus groups attached to the triple
bond are constrained to the shielding region and are shifted upfield relative
to similar vinyl protons. Thus acetylenic protons come at δ = 2 – 3 and
propargylic methyl groups are upfield from allylic methyl groups.
The
chemical shift of a given proton is thus determined by a combina-tion of
isotropic shielding by the electron cloud surrounding the proton and by
anisotropic shielding due to the presence of nearby functional groups which are
strongly anisotropic. These factors are usually sufficient to give unique
chemical shifts for most protons in a molecule, and they can normally be
distinguished using modern high-field NMR spectrometers (200 – 300 MHz).
Furthermore the integration of these signals gives the numbers of the different
types of protons.
Related Topics
