Calcium Channel Blockers
| Home | | Pharmacology |Chapter: Essential pharmacology : Antianginal and Other Anti-Ischaemic Drugs
Verapamil was developed in Germany in 1962 as a coronary dilator. It had additional cardio-depressant property, but its mechanism of action was not known. Fleckenstein (1967) showed that it interfered with Ca2+ movement into the cell.
CALCIUM CHANNEL BLOCKERS
Verapamil was developed in Germany in 1962 as a coronary dilator. It had additional cardio-depressant property, but its mechanism of action was not known. Fleckenstein (1967) showed that it interfered with Ca2+ movement into the cell. In the subsequent years, a large number of chemically diverse Ca2+ channel blockers (CCBs) with different pharmacological profiles have been produced.
Three important classes of calcium channel blockers are examplified by:
Verapamil—a phenyl alkylamine, hydrophilic papaverine congener.
Nifedipine—a dihydropyridine (lipophilic).
Diltiazem—a hydrophilic benzothiazepine.
The dihydropyridines (DHPs) are the most potent Ca2+ channel blockers, and this subclass has proliferated exceptionally.
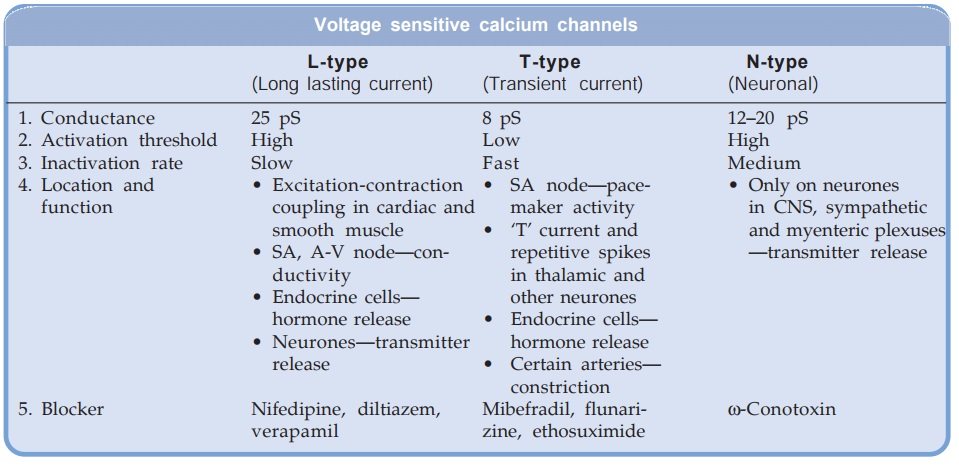
Calcium Channels
Three types of Ca2+ channels have been described in smooth muscles (other excitable cells as well):
(a) Voltage Sensitive Channel Activated when membrane potential drops to around –40 mV or lower.
(b) Receptor Operated Channel Activated by Adr and other agonists—independent of membrane depolarization (NA contracts even depolarized aortic smooth muscle by promoting influx of Ca2+ through this channel and releasing Ca2+ from sarcoplasmic reticulum).
(c) Leak Channel Small amounts of Ca2+ leak into the resting cell and are pumped out by Ca2+ATPase.
Mechanical stretch promotes inward movement of Ca2+, which may be occurring through activation of the leak channel or through separate stretch sensitive channel.
The voltage sensitive Ca2+ channels are heterogeneous: three major types have been identified.
All voltage sensitive Ca2+ channels are membrane spanning funnel shaped glycoproteins that function as ion selective valves. They are composed of a major α1 subunit which encloses the ion channel and other modulatory subunits like α2,β, γ and δ. In L-type Ca2+ channels each subunit exists in multiple isoforms which may be site specific, e.g.
Skeletal muscle L-channels are: α1s . α2/δa . β1 . γ
Cardiac muscle L-channels are: α1ca . α2/δc . β2
Smooth muscle L-channels are: α1cb . α2/δ . β3
Even smooth muscle L-channels differ between vascular and nonvascular. Moreover distribution may be heterogeneous in different parts of the vascular bed.
Only the voltage sensitive L-type channels are blocked by the CCBs. The 3 groups of CCBs viz. phenylalkylamines (verapamil), benzothiazepine (diltiazem) and dihydropyridines (nifedipine) bind to their own specific binding sites on the α1 subunit; all restricting Ca2+ entry, though characteristics of channel blockade differ. Further, different drugs may have differing affinities for various site specific isoforms of the L-channels. This may account for the differences in action exhibited by various CCBs. The vascular smooth muscle has a more depolarized membrane (RMP about –40 mV) than heart. This may contribute to vascular selectivity of certain CCBs.
Pharmacological Actions And Adverse Effects
The common property of all three subclasses of CCBs is to inhibit Ca2+ mediated slow channel component of action potential (AP) in smooth/ cardiac muscle cell. The two most important actions of CCBs are:
· Smooth muscle (especially vascular) relaxation.
· Negative chronotropic, inotropic and dromotropic action on heart.
Smooth Muscle
Smooth muscles depolarise primarily by inward Ca2+ movement through voltage sensitive channel. These Ca2+ ions trigger release of more Ca2+ from intracellular stores and together bring about excitation contraction coupling through phosphorylation of myosin light chain as depicted in Fig. 39.3. CCBs cause relaxation by decreasing intracellular availability of Ca2+. They markedly relax arterioles but have mild effect on veins. Extravascular smooth muscle (bronchial, biliary, intestinal, vesical, uterine) is also relaxed.
The dihydropyridines (DHPs) have the most marked smooth muscle relaxant and vasodilator action; verapamil is somewhat weaker followed by diltiazem.
Nitrendipine and other DHPs have been shown to release NO from endothelium and inhibit cAMP-phosphodiesterase resulting in raised smooth muscle cAMP. These additional mechanisms may account for their predominant smooth muscle relaxant action. Released endothelial NO may exert anti-atherosclerotic action.
Heart In the working atrial and ventricular fibres, Ca2+ moves in during plateau phase of AP →releases more Ca2+ from sarcoplasmic reticulum →contraction through binding to troponin— allowing interaction of myosin with actin. The CCBs would thus have negative inotropic action.
The 0 phase depolarization in SA and AV nodes is largely Ca2+ mediated. Automaticity and conductivity of these cells appear to be dependent on the rate of recovery of the Ca2+ channel.
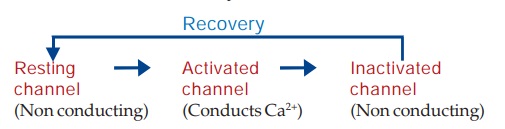
The L-type Ca2+ channels activate as well as inactivate at a slow rate. Consequently, Ca2+ depolarized cells (SA and AV nodal) have a considerably less steep 0 phase and longer refractory period. The recovery process which restores the channel to the state from which it can again be activated by membrane depolarization is delayed by verapamil and to a lesser extent by diltiazem (resulting in depression of pacemaker activity and conduction), but not by DHPs (they have no negative chronotropic/dromotropic action). Moreover, channel blockade by verapamil is enhanced at higher rates of stimulation, that by nifedipine is independent of frequency, while diltiazem is intermediate. Thus, verapamil slows sinus rate and AV conduction, but nifedipine does not. Effect of diltiazem on sinus node automaticity and AV conduction is similar to that of verapamil.
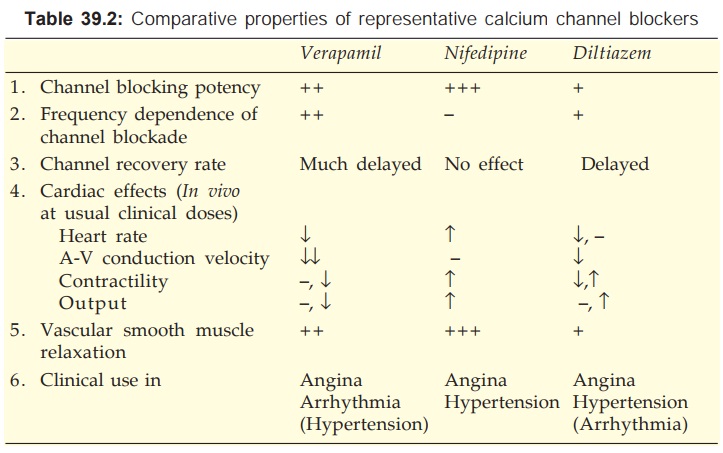
The relative potencies to block slow channels in smooth muscle do not parallel those in the heart. The DHPs are more selective for smooth muscle L-channels: at concentrations which cause vasodilatation they have negligible negative inotropic action. Diltiazem causes less depression of contractility than verapamil. Important differences between the three representative CCBs are summarized in Table 39.2. Their cardiac electrophysiological effects are compared in Table 38.1.
Verapamil
It dilates arterioles and has some α adrenergic blocking activity—decreases t.p.r. but BP is only modestly lowered. The pronounced direct cardio-depressant effect is partially offset in vivo by reflex effects of peripheral vasodilatation. The HR generally decreases, AV conduction is slowed, but c.o. is maintained by reflex sympathetic stimulation and reduction in aortic impedance. However, ventricular contractility may be markedly impaired in CHF patients. Coronary flow is increased.
Dose: 40–160 mg TDS oral, 5 mg by slow i.v. injection. CALAPTIN 40, 80 mg tabs, 120, 240 mg SR tabs, 5 mg/ 2 ml inj.
Adverse Effects
Nausea, constipation and bradycardia are more common than other CCBs, while flushing, headache and ankle edema are less common. Hypotension is occasional and tachycardia (common with DHPs) is absent. It can accentuate conduction defects (contraindicated in 2nd and 3rd degree AV block) and precipitate CHF in patients with preexisting disease. Cardiac arrest has occurred on i.v. injection and when it is given to patients with sick sinus.
Interactions
Verapamil should not be given with β blockers—additive sinus depression, conduction defects or asystole may occur.
It increases plasma digoxin level by decreasing its excretion: toxicity can develop.
It should not be used with other cardiac depressants like quinidine and disopyramide.
Diltiazem
It is a less potent vasodilator than nifedipine and verapamil, and has modest direct negative inotropic action, but direct depression of SA node and AV conduction are equivalent to verapamil. Usual clinical doses produce consistent fall in BP with little change or decrease in HR. Large dose or i.v. injection decreases t.p.r. markedly which may elicit reflex cardiac effects. It dilates coronaries.
Dose: 30–60 mg TDS–QID oral; DILZEM, 30, 60 mg tabs, 90 mg SR tab; 25 mg/5 ml inj; ANGIZEM 30, 60, 90, 120, 180 mg tab, DILTIME 30, 60 mg tab; 90, 120 mg SR tab.
Adverse Effects
Incidence of side effects is low, but the profile is similar to verapamil. Like verapamil, it also increases plasma digoxin.
Diltiazem should not be given to patients with preexisting sinus, AV nodal or myocardial disease. Only low doses should be given to patients on β blockers.
Nifedipine
It is the prototype DHP with a rapid onset and short duration of action. The overriding action of nifedipine is arteriolar dilatation → t.p.r. decreases, BP falls. The direct depressant effect on heart requires much higher dose, but a weak negative inotropic action can be unmasked after β blockade. As discussed above, it does not depress SA node or AV conduction. Reflex sympathetic stimulation of heart predominates → tachycardia, increased contractility and c.o. (no decrease in venous return along with lowering of afterload aid increase in c.o.). Coronary flow is increased.
Nifedipine has mild natriuretic action, but significant diuresis does not occur.
Dose: 5–20 mg BD–TDS oral.
CALCIGARD, DEPIN, NIFELAT 5, 10 mg tab, also 10 mg, 20 mg S.R. (RETARD) tab; NICARDIA 5, 10 mg tab; 10, 20, 30 mg SR tab.
Adverse Effects
Frequent side effects are palpitation, flushing, ankle edema, hypotension, headache, drowsiness and nausea. These are related to peaks of drug level in blood: can be minimized by low starting dose, fractionation of dose or use of retard formulation. Nifedipine has paradoxically increased the frequency of angina in some patients. Higher mortality among post MI patients has been confirmed. However, it has been safely administered with β blockers and digoxin.
By its relaxant effect on bladder nifedipine can increase urine voiding difficulty in elderly males. It has also been reported to hamper diabetes control by decreasing insulin release.
OTHER DIHYDROPYRIDINES (DHPS)
All DHPs have pharmacodynamic profile similar to nifedipine; there are minor differences in organ selectivity and major differences in pharmaco kinetic characteristics. The slower and longer acting ones induce less reflex sympathetic stimulation. Tachycardia, propensity to increase cardiac work, flushing, headache, dizziness are subdued. They are currently favoured, particularly since increased mortality among postMI patients has been reported with the regular short-acting nifedipine formulation.
Felodipine
It differs from nifedipine in having greater vascular selectivity, larger tissue distribution and longer t½. The extended release preparation is suitable for once daily administration.
Dose: 5–10 mg OD, max. 10 mg BD.
FELOGARD, PLENDIL, RENDIL 2.5, 5, 10 mg ER tab.
Amlodipine
Pharmacokinetically it is the most distinct DHP. It has complete but slow oral absorption: peak after 6 to 9 hr—the early vasodilator side effects (palpitation, flushing, headache, postural dizziness) are largely avoided. Because of less extensive and less variable first pass metabolism, its oral bioavailability is higher and more consistent. Volume of distribution and t½ are exceptionally long: diurnal fluctuation in blood level is small and action extends over the next morning.
Dose: 5–10 mg OD; AMLOPRES, AMCARD, AMLOPIN, MYODURA 2.5, 5, 10 mg tabs.
S(–)Amlodipine The single enantiomer preparation is effective at half the dose and is claimed to cause less ankle edema.
Dose: 2.5–5 mg OD;
SNUMLO, SAMCARD, ASOMEX, ESAM 2.5, 5 mg tabs.
Nitrendipine
A DHP with oral bioavailability of 10–30% and elimination t½ of 4–12 hours. It has been shown to release NO from the endothelium and inhibit cAMP phosphodiesterase; which may be the additional mechanisms of vasodilator action. The endothelial NO is claimed to retard atherosclerosis. Ventricular contractility and AV conduction are not depressed. Nitrendipine is indicated in hypertension and angina pectoris.
Dose: 5–20 mg OD; NITREPIN, CARDIF 10, 20 mg tabs.
Lacidipine
A highly vasoselective newer DHP suitable for once daily administration. It is claimed to attain higher concentration in vascular smooth muscle membrane; approved only for use as antihypertensive.
Dose: 4 mg OD, increase to 6 mg OD if required.
LACIVAS, SINOPIL 2, 4 mg tabs.
Nimodipine
It is a short-acting DHP which penetrates bloodbrain barrier very efficiently due to high lipid solubility. It selectively relaxes cerebral vasculature; approved for prevention and treatment of neurological deficit due to cerebral vasospasm following subarachnoid haemorrhage or ruptured congenital intracranial aneurysms. Side effects are headache, flushing, dizziness, palpitation and nausea.
Dose: 30–60 mg 4–6 hourly for 3 weeks following subarachnoid haemorrhage; VASOTOP, NIMODIP, NIMOTIDE 30 mg tab; 10 mg/50 ml inj.
Lercanidipine
Another DHP similar to nifedipine, but with longer duration of action. Peak plasma concentrations occur at 1.5–3 hrs; t½ is 5–10 hours. It is indicated in hypertension at a dose of 10–20 mg OD.
LEREZ, LERKA 10, 20 mg tabs.
Benidipine
A long-acting DHP that owes its long duration of action to slow dissociation from the DHP receptor on the smooth muscle cell. It is indicated in hypertension and angina pectoris. It is marketed only in India and Japan.
Dose: 4–8 mg OD; CARITEC 4, 8 mg tab.
Pharmacokinetics
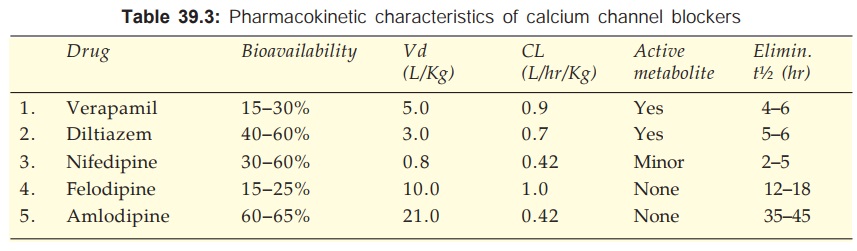
The pharmacokinetic parameters of Ca2+ channel blockers are tabulated in Table 39.3. All are 90–100% absorbed orally, peak occurring at 1–3 hr (except amlodipine 6–9 hr). The oral bioavailability of Ca2+ channel blockers is incomplete with marked inter and intra individual variations. This is due to high first pass metabolism (modest and less variable for amlodipine). All are highly plasma protein bound (min.: diltiazem 80%, max.: felodipine 99%).
The Ca2+ channel blockers are high clearance drugs with extensive tissue distribution. All are 90% metabolized in liver and excreted in urine. Some metabolites are active. The elimination t½ are in the range of 2–6 hr, but that of amlodipine is exceptionally long; followed by lacidipine, nitrendipine and felodipine.
On chronic use verapamil decreases its own metabolism—bioavailability is nearly doubled and t½ is prolonged.
Uses
Calcium channel blockers can be safely given to patients with obstructive lung disease and peripheral vascular disease in whom β blockers are contraindicated. The problem of rebound worsening of angina on withdrawal after chronic use is less with CCBs than with β blockers.
Angina Pectoris
All CCBs are effective in reducing frequency and severity of classical as well as variant angina. Benefit in classical angina appears to be primarily due to reduction in cardiac work: mainly as a result of reduced afterload. Though, they can increase coronary flow in normal individuals, this is unlikely to be significant in patients with fixed arterial obstruction. Exercise tolerance is increased.
Many controlled studies and meta-analysis have concluded that myocardial ischaemia may be aggravated by short-acting DHPs. This may be due to decreased coronary flow secondary to fall in mean arterial pressure, reflex tachycardia and coronary steal. The direct cardiac effect of verapamil and diltiazem to reduce O2 requirement and less marked sympathetic stimulation makes them less likely to aggravate ischaemia.
Trials using high dose regular short-acting nifedipine formulation have reported increased mortality among MI patients. The sudden rush of sympathetic activity evoked by each dose of these preparations has been held responsible for the deleterious effect. The slow and long-acting DHPs do not share this disadvantage. There is some evidence that verapamil and diltiazem reduce reinfarction and mortality in MI patients (equal to that achieved by β blockers) with uncompromised ventricular function.
Myocardial infarction: The concensus opinion is against use of CCBs in MI, but verapamil/ diltiazem may be employed for secondary prophylaxis when β blockers are contraindicated.
The capacity of CCBs to prevent arterial spasm is undoubtedly responsible for the beneficial effect in variant angina. Reduction of cardiac O2 demand would also work in the same direction. No significant difference in efficacy among different CCBs has been noted in angina pectoris.
CCBs are not a first line treatment of unstable angina; may be used as add on therapy to nitrates when coronary vasospasm is prominent and is not counteracted by nitrate alone. Use of nifedipine/DHPs in non β blocked patients is to be avoided.
Hypertension
DHPs, diltiazem and verapamil are among the first line drugs for hypertension (see Ch. No. 40).
Cardiac Arrhythmias
Verapamil and diltiazem are highly effective in PSVT and for control of ventricular rate in supraventricular arrhythmias (see Ch. No. 38).
Hypertrophic Cardiomyopathy
The negative inotropic action of verapamil can be salutary in this condition.
Other Uses
Nifedipine is an alternative drug for premature labour; Verapamil has been used to suppress migraine and nocturnal leg cramps. The DHPs reduce severity of Raynaud’s episodes.
Related Topics
